| A549-all.twopass.merge150.wgt10.zgt2.wig | Combined | DNase-seq | ENCODE | A549 | Epithelium | Hotspots | A549 | NA | GSM816649,
GSM736580,
GSM736506 |
Adult_Th0_AllReps.30000000.twopass.merge150.wgt10.
zgt2.wig | UW | DNase-seq | ENCODE | Adult_CD4+ | Blood | Hotspots | Adult_CD4+ | NA | Not found |
| AG04449-DS12319.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | AG04449 | Skin | Hotspots | AG04449 | NA | GSM736562,
GSM736590 |
| AG04450-DS12270.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | AG04450 | Lung | Hotspots | AG04450 | NA | GSM736514,
GSM736563 |
| AG09309-DS12352.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | AG09309 | Skin | Hotspots | AG09309 | NA | GSM736551,
GSM736616 |
| AG09319-DS12291.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | AG09319 | Gingival | Hotspots | AG09319 | NA | GSM736531,
GSM736619 |
| AG10803-DS12384.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | AG10803 | Skin | Hotspots | AG10803 | NA | GSM736598,
GSM736633 |
| AoAF-DS13523.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | AoAF | Blood vessel | Hotspots | AoAF | NA | GSM736583,
GSM736505 |
| BE_2_C-DS14625.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | BE2_C | Brain | Hotspots | BE2_C | NA | Not found |
| BJ-DS10081.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | BJ | Skin | Hotspots | BJ | NA | GSM736518,
GSM736596 |
| CACO2-DS8235.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | Caco-2 | Colon | Hotspots | Caco-2 | NA | GSM736500,
GSM736587 |
| CD14-DS18065.hg19.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | CD14+ | Blood | Hotspots | CD14+ | NA | Not found |
| CD20-DS17541.hg19.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | CD20+ | Blood | Hotspots | CD20+ | NA | Not found |
| CD34-DS16814.hg19.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | CD34+ | Blood | Hotspots | CD34+ | NA | Not found |
| CMK-DS12393.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | CMK | Blood | Hotspots | CMK | NA | GSM736607 |
E_myoblast_AllReps.30000000.twopass.merge150.
wgt10.zgt2.wig | UW | DNase-seq | ENCODE | E_myoblast | Muscle | Hotspots | E_myoblast | NA | Not found |
| GM06990-DS7748.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | GM06990 | Blood | Hotspots | GM06990 | NA | GSM736558,
GSM736635 |
| GM12864-DS12431.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | GM12864 | Blood | Hotspots | GM12864 | NA | GSM736525 |
| GM12865-DS12436.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | GM12865 | Blood | Hotspots | GM12865 | NA | GSM736512,
GSM736561 |
| GM12878-all.twopass.merge150.wgt10.zgt2.wig | Combined | DNase-seq | ENCODE | GM12878 | Blood | Hotspots | GM12878 | NA | GSM816665,
GSM736496,
GSM736620 |
| HAc-DS14765.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HAc | Cerebellar | Hotspots | HAc | NA | GSM736586,
GSM736538 |
| HAEpiC-DS12663.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HAEpiC | Epithelium | Hotspots | HAEpiC | NA | GSM736631,
GSM736606 |
| HAh-DS15192.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HA-h | Brain hippocampus | Hotspots | HA-h | NA | GSM736594,
GSM736535 |
| HAsp-DS14790.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HA-sp | Spinal cord | Hotspots | HA-sp | NA | GSM736537,
GSM736625 |
| HBMEC-DS13817.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HBMEC | Blood vessel | Hotspots | HBMEC | NA | GSM736509,
GSM736554 |
| HCFaa-DS13480.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HCFaa | Heart | Hotspots | HCFaa | NA | GSM736494,
GSM736601 |
| HCF-DS12501.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HCF | Heart | Hotspots | HCF | NA | GSM736568,
GSM736540 |
| HCM-DS12599.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HCM | Heart | Hotspots | HCM | NA | GSM736516,
GSM736504 |
| HConF-DS11642.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HConF | Eye | Hotspots | HConF | NA | GSM736547,
GSM736515 |
| HCPEpiC-DS12447.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HCPEpiC | Epithelium | Hotspots | HCPEpiC | NA | GSM736569,
GSM736597 |
| HCT116-DS13551.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HCT-116 | Colon | Hotspots | HCT-116 | NA | GSM736600,
GSM736493 |
| HEEpiC-DS12763.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HEEpiC | Epithelium | Hotspots | HEEpiC | NA | GSM736585,
GSM736532 |
| Hela-all.twopass.merge150.wgt10.zgt2.wig | Combined | DNase-seq | ENCODE | HeLa-S3 | Cervix | Hotspots | HeLa-S3 | NA | GSM816633,
GSM816643,
GSM736564,
GSM736510 |
| HepG2-all.twopass.merge150.wgt10.zgt2.wig | Combined | DNase-seq | ENCODE | HepG2 | Liver | Hotspots | HepG2 | NA | GSM816662,
GSM736637,
GSM736639 |
| HESC-all.twopass.merge150.wgt10.zgt2.wig | Combined | DNase-seq | ENCODE | HESC | ES Cell | Hotspots | HESC | NA | Not found |
| hESCT0-DS11909.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | hESCT0 | ES Cell | Hotspots | hESCT0 | NA | Not found |
| HFF-DS15115.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HFF | Foreskin | Hotspots | HFF | NA | GSM736602,
GSM736572 |
| HFF_MyC-DS15079.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HFF-Myc | Foreskin | Hotspots | HFF-Myc | NA | GSM736524,
GSM736605 |
| HGF-DS11752.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HGF | Gingival | Hotspots | HGF | NA | GSM736579,
GSM736576 |
| HIPEpiC-DS12684.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HIPEpiC | Epithelium | Hotspots | HIPEpiC | NA | GSM736589,
GSM736615 |
| HL60-DS11809.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HL-60 | Blood | Hotspots | HL-60 | NA | GSM736626,
GSM736595 |
| HMEC-all.twopass.merge150.wgt10.zgt2.wig | Combined | DNase-seq | ENCODE | HMEC | Breast | Hotspots | HMEC | NA | GSM816669,
GSM736634,
GSM736552 |
| HMF-DS13363.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HMF | Breast | Hotspots | HMF | NA | GSM736628,
GSM736541 |
HMVEC_dAd-DS12957.hg19.twopass.merge150.wgt10.
zgt2.wig | UW | DNase-seq | ENCODE | HMVEC-dAd | Blood vessel | Hotspots | HMVEC-dAd | NA | GSM1024745,
GSM1024747 |
HMVEC_dBlAd-DS13337.twopass.merge150.wgt10.
zgt2.wig | UW | DNase-seq | ENCODE | HMVEC-dBl-Ad | Blood vessel | Hotspots | HMVEC-dBl-Ad | NA | GSM736609,
GSM736523 |
HMVEC_dBlNeo-DS13242.twopass.merge150.wgt10.
zgt2.wig | UW | DNase-seq | ENCODE | HMVEC-dBl-Neo | Blood vessel | Hotspots | HMVEC-dBl-Neo | NA | GSM736571,
GSM736521 |
HMVEC_dLyAd-DS13261.twopass.merge150.wgt10.
zgt2.wig | UW | DNase-seq | ENCODE | HMVEC-dLy-Ad | Blood vessel | Hotspots | HMVEC-dLy-Ad | NA | GSM736599,
GSM736591 |
HMVEC_dLyNeo-DS13150.twopass.merge150.wgt10.
zgt2.wig | UW | DNase-seq | ENCODE | HMVEC-dLy-Neo | Blood vessel | Hotspots | HMVEC-dLy-Neo | NA | GSM736577,
GSM736573 |
| HMVEC_dNeo-DS12937.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HMVEC-dNeo | Blood vessel | Hotspots | HMVEC-dNeo | NA | GSM736611,
GSM736624 |
| HMVEC_LBl-DS13372.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HMVEC-LBl | Blood vessel | Hotspots | HMVEC-LBl | NA | GSM736542,
GSM736511 |
| HMVEC_LLy-DS13185.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HMVEC-LLy | Blood vessel | Hotspots | HMVEC-LLy | NA | GSM736507,
GSM736627 |
| HNPCEpiC-DS12467.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HNPCEpiC | Epithelium | Hotspots | HNPCEpiC | NA | GSM736621,
GSM736550 |
| HPAEC-DS12916.hg19.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HPAEC | Blood vessel | Hotspots | HPAEC | NA | GSM1024763 |
| HPAF-DS13411.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HPAF | Blood vessel | Hotspots | HPAF | NA | GSM736555,
GSM736614 |
| HPdLF-DS13573.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HPdLF | Epithelium | Hotspots | HPdLF | NA | GSM736632,
GSM736528 |
| HPF-DS13390.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HPF | Lung | Hotspots | HPF | NA | GSM736574,
GSM736503 |
| HRCE-DS10666.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HRCEpiC | Epithelium | Hotspots | HRCEpiC | NA | GSM736549,
GSM736557 |
| HRE-DS10641.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HRE | Epithelium | Hotspots | HRE | NA | GSM736527,
GSM736548 |
| HRGEC-DS13716.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HRGEC | Kidney | Hotspots | HRGEC | NA | GSM736499,
GSM736618 |
| HRPEpiC-DS12583.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HRPEpiC | Epithelium | Hotspots | HRPEpiC | NA | GSM736630,
GSM736623 |
| HSMM-all.twopass.merge150.wgt10.zgt2.wig | Combined | DNase-seq | ENCODE | HSMM | Muscle | Hotspots | HSMM | NA | GSM816650,
GSM736560,
GSM736553 |
| HSMM_D-all.twopass.merge150.wgt10.zgt2.wig | Combined | DNase-seq | ENCODE | HSMM | Muscle | Hotspots | HSMM | NA | GSM816650,
GSM736560,
GSM736553 |
| hTH1-all.twopass.merge150.wgt10.zgt2.wig | Combined | DNase-seq | ENCODE | Th1 | Blood | Hotspots | Th1 | NA | GSM736592,
GSM1024760 |
| hTH2-DS7842.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | Th2 | Blood | Hotspots | Th2 | NA | GSM736502,
GSM1024792 |
| HUVEC-all.twopass.merge150.wgt10.zgt2.wig | Combined | DNase-seq | ENCODE | HUVEC | Blood vessel | Hotspots | HUVEC | NA | GSM816646,
GSM736575,
GSM736533 |
| HVMF-DS13981.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | HVMF | Connective | Hotspots | HVMF | NA | GSM736534,
GSM736491 |
Ishikawa_E_AllReps.30000000.twopass.merge150.
wgt10.zgt2.wig | UW | DNase-seq | ENCODE | Ishikawa | Uterus | Hotspots | Ishikawa | NA | GSM1008594,
GSM1008593 |
Ishikawa_T_AllReps.30000000.twopass.merge150.
wgt10.zgt2.wig | UW | DNase-seq | ENCODE | Ishikawa | Uterus | Hotspots | Ishikawa | NA | GSM1008594,
GSM1008593 |
| Jurkat-DS12659.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | Jurkat | Blood | Hotspots | Jurkat | NA | GSM736501,
GSM736492 |
| K562-all.twopass.merge150.wgt10.zgt2.wig | Combined | DNase-seq | ENCODE | K562 | Blood | Hotspots | K562 | NA | GSM816655,
GSM1008602,
GSM1008567,
GSM1008601,
GSM1008558,
GSM1008580,
GSM736629,
GSM736566 |
| LNCap-all.twopass.merge150.wgt10.zgt2.wig | Combined | DNase-seq | ENCODE | LNCaP | Prostate | Hotspots | LNCaP | NA | GSM816637,
GSM816634,
GSM736565,
GSM736603 |
| MCF7-all.twopass.merge150.wgt10.zgt2.wig | Combined | DNase-seq | ENCODE | MCF-7 | Breast | Hotspots | MCF-7 | NA | GSM816627,
GSM1008581,
GSM816670,
GSM1008565,
GSM1008603,
GSM1024784,
GSM1024783,
GSM1024764,
GSM1024767,
GSM736581,
GSM736588 |
| NB4-DS12543.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | NB4 | Blood | Hotspots | NB4 | NA | GSM736604,
GSM736529 |
| NHA-DS12800.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | NH-A | Nervous | Hotspots | NH-A | NA | GSM736544,
GSM736584 |
| NHDF_Ad-DS12863.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | NHDF-Ad | Skin | Hotspots | NHDF-Ad | NA | GSM736567,
GSM736520 |
| NHDF_Neo-DS11923.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | NHDF-neo | Skin | Hotspots | NHDF-neo | NA | GSM736498,
GSM736546 |
| NHEK-all.twopass.merge150.wgt10.zgt2.wig | Combined | DNase-seq | ENCODE | NHEK | Skin | Hotspots | NHEK | NA | GSM816635,
GSM736545,
GSM736556 |
| NHLF-DS12829.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | NHLF | Lung | Hotspots | NHLF | NA | GSM736612,
GSM736536 |
| NT2_D1-DS14575.hg19.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | NT2-D1 | Testis | Hotspots | NT2-D1 | NA | GSM1024751,
GSM1024795 |
| PANC1-DS9955.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | PANC-1 | Pancreas | Hotspots | PANC-1 | NA | GSM736517,
GSM736519 |
| PrEC-DS12098.hg19.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | PrEC | Prostate | Hotspots | PrEC | NA | GSM1024742,
GSM1024743 |
| RPTEC-DS14061.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | RPTEC | Epithelium | Hotspots | RPTEC | NA | GSM736543,
GSM736539 |
RWPE_AllReps.30000000.twopass.merge150.wgt10.
zgt2.wig | UW | DNase-seq | ENCODE | RWPE1 | Prostate | Hotspots | RWPE1 | NA | GSM1008595 |
| SAEC-DS10518.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | SAEC | Epithelium | Hotspots | SAEC | NA | GSM736608,
GSM736617 |
| SkMC-DS11949.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | SKMC | Muscle | Hotspots | SKMC | NA | Not found |
| SK_N_MC-DS14408.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | SK-N-MC | Brain | Hotspots | SK-N-MC | NA | GSM736522,
GSM736570 |
| SKNSH-DS8482.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | SK-N-SH | Brain | Hotspots | SK-N-SH | NA | GSM1008585 |
| WERI_Rb1-DS13681.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | ENCODE | WERI-Rb-1 | Eye | Hotspots | WERI-Rb-1 | NA | GSM736495,
GSM736636 |
wgEncodeOpenChromDnase8988tAlnAllReps.30000000.
twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | 8988T | Liver | Hotspots | 8988T | NA | GSM816667 |
wgEncodeOpenChromDnaseAosmcSerumfreeAlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | AoSMC | Blood vessel | Hotspots | AoSMC | NA | GSM816638 |
wgEncodeOpenChromDnaseChorionAlnAllReps.30000000.
twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | Chorion | Fetal
membrane | Hotspots | Chorion | NA | GSM816628 |
wgEncodeOpenChromDnaseCllAlnAllReps.30000000.
twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | CLL | Blood | Hotspots | CLL | NA | GSM816664 |
wgEncodeOpenChromDnaseFibroblAlnAllReps.30000000.
twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | Fibrobl | Skin | Hotspots | Fibrobl | NA | GSM816652 |
wgEncodeOpenChromDnaseFibropAlnAllReps.30000000.
twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | FibroP | Skin | Hotspots | FibroP | NA | GSM816626 |
wgEncodeOpenChromDnaseGlioblaAlnAllReps.30000000.
twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | Gliobla | Brain | Hotspots | Gliobla | NA | GSM816668 |
wgEncodeOpenChromDnaseGm12891AlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | GM12891 | Blood | Hotspots | GM12891 | NA | GSM816656 |
wgEncodeOpenChromDnaseGm12892AlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | GM12892 | Blood | Hotspots | GM12892 | NA | GSM816657 |
wgEncodeOpenChromDnaseGm18507AlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | GM18507 | Blood | Hotspots | GM18507 | NA | GSM816653 |
wgEncodeOpenChromDnaseGm19238AlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | GM19238 | Blood | Hotspots | GM19238 | NA | GSM816658 |
wgEncodeOpenChromDnaseGm19239AlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | GM19239 | Blood | Hotspots | GM19239 | NA | GSM816659 |
wgEncodeOpenChromDnaseGm19240AlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | GM19240 | Blood | Hotspots | GM19240 | NA | GSM816648 |
wgEncodeOpenChromDnaseH9esAlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | H9ES | ES Cell | Hotspots | H9ES | NA | GSM816629 |
wgEncodeOpenChromDnaseHelas3Ifna4hAlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | HeLa-S3 | Cervix | Hotspots | HeLa-S3 | NA | GSM816633,
GSM816643,
GSM736564,
GSM736510 |
wgEncodeOpenChromDnaseHepatocytesAlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | Hepatocytes | Liver | Hotspots | Hepatocytes | NA | GSM816663 |
wgEncodeOpenChromDnaseHpde6e6e7AlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | HPDE6-E6E7 | Pancreatic
duct | Hotspots | HPDE6-E6E7 | NA | GSM816639 |
wgEncodeOpenChromDnaseHtr8AlnAllReps.30000000.
twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | HTR8svn | Blastula | Hotspots | HTR8svn | NA | GSM816644 |
wgEncodeOpenChromDnaseHuh75AlnAllReps.30000000.
twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | Huh-7.5 | Liver | Hotspots | Huh-7.5 | NA | GSM816671 |
wgEncodeOpenChromDnaseHuh7AlnAllReps.30000000.
twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | Huh-7 | Liver | Hotspots | Huh-7 | NA | GSM816641 |
wgEncodeOpenChromDnaseIpsAlnAllReps.30000000.
twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | iPS | IPS | Hotspots | iPS | NA | GSM816642 |
wgEncodeOpenChromDnaseLncapAndroAlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | LNCaP | Prostate | Hotspots | LNCaP | NA | GSM816637,
GSM816634,
GSM736565,
GSM736603 |
wgEncodeOpenChromDnaseMcf7HypoxlacAlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | MCF-7 | Breast | Hotspots | MCF-7 | NA | GSM816627,
GSM1008581,
GSM816670,
GSM1008565,
GSM1008603,
GSM1024784,
GSM1024783,
GSM1024764,
GSM1024767,
GSM736581,
GSM736588 |
wgEncodeOpenChromDnaseMedulloAlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | Medullo | Brain | Hotspots | Medullo | NA | GSM816636 |
wgEncodeOpenChromDnaseMelanoAlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | Melano | Skin | Hotspots | Melano | NA | GSM816631 |
wgEncodeOpenChromDnaseMyometrAlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | Myometr | Myometrium | Hotspots | Myometr | NA | GSM816630 |
wgEncodeOpenChromDnaseOsteoblAlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | Osteobl | Bone | Hotspots | Osteobl | NA | Not found |
wgEncodeOpenChromDnasePanisdAlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | PanIsletD | Pancreas | Hotspots | PanIsletD | NA | GSM816666 |
wgEncodeOpenChromDnasePanisletsAlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | PanIslets | Pancreas | Hotspots | PanIslets | NA | GSM816660 |
wgEncodeOpenChromDnasePhteAlnAllReps.30000000.
twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | pHTE | Epithelium | Hotspots | pHTE | NA | GSM816647 |
wgEncodeOpenChromDnaseProgfibAlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | ProgFib | Skin | Hotspots | ProgFib | NA | GSM816661 |
wgEncodeOpenChromDnaseStellateAlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | Stellate | Liver | Hotspots | Stellate | NA | GSM816672 |
wgEncodeOpenChromDnaseT47dAlnAllReps.30000000.
twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | T-47D | Breast | Hotspots | T-47D | NA | GSM816673,
GSM1008576,
GSM1024762,
GSM1024761 |
wgEncodeOpenChromDnaseUrotsaAlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | Urothelia | Urothelium | Hotspots | Urothelia | NA | GSM1008606,
GSM1008605 |
wgEncodeOpenChromDnaseUrotsaUt189AlnAllReps.
30000000.twopass.merge150.wgt10.zgt2.wig | Duke:
UNC:UTA | DNase-seq | ENCODE | Urothelia | Urothelium | Hotspots | Urothelia | NA | GSM1008606,
GSM1008605 |
| WI_38-DS14315.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | WI-38 | Embryonic
lung | Hotspots | WI-38 | NA | GSM736613,
GSM736526,
GSM736613,
GSM736526 |
| WI_38_TAM-DS14323.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | WI-38 | Embryonic
lung | Hotspots | WI-38 | NA | GSM736613,
GSM736526,
GSM736613,
GSM736526 |
UW.Breast_vHMEC.ChromatinAccessibility.RM035.
DS18406.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Breast_vHMEC.
RM035 | Breast | Hotspots | Breast_vHMEC | RM035 | GSM753973 |
UW.Breast_vHMEC.ChromatinAccessibility.RM035.
DS18438.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Breast_vHMEC.
RM035 | Breast | Hotspots | Breast_vHMEC | RM035 | GSM753974 |
UW.CD14_Primary_Cells.ChromatinAccessibility.
RO_01689.DS17391.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD14_Primary_
Cells.RO_01689+ | Blood | Hotspots | CD14 | RO_01689+ | GSM701503 |
UW.CD14_Primary_Cells.ChromatinAccessibility.
RO_01701.DS17889.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD14_Primary_
Cells.RO_01701+ | Blood | Hotspots | CD14 | RO_01701+ | GSM701541 |
UW.CD19_Primary_Cells.ChromatinAccessibility.
RO_01679.DS17186.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD19_Primary_
Cells.RO_01679+ | Blood | Hotspots | CD19 | RO_01679+ | GSM701492 |
UW.CD19_Primary_Cells.ChromatinAccessibility.
RO_01689.DS17281.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD19_Primary_
Cells.RO_01689+ | Blood | Hotspots | CD19 | RO_01689+ | GSM701493 |
UW.CD19_Primary_Cells.ChromatinAccessibility.
RO_01701.DS17440.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD19_Primary_
Cells.RO_01701+ | Blood | Hotspots | CD19 | RO_01701+ | GSM701507 |
UW.CD20_Primary_Cells.ChromatinAccessibility.
RO_01778.DS17371.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD20_Primary_
Cells.RO_01778+ | Blood | Hotspots | CD20 | RO_01778+ | GSM701500 |
UW.CD3_Primary_Cells.ChromatinAccessibility.CB1_1-3-
2011.DS17702.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD3_Primary_
Cells.CB1_1-3-
2011+ | Blood | Hotspots | CD3 | CB1_1-3-
2011+ | GSM701525 |
UW.CD3_Primary_Cells.ChromatinAccessibility.
CB6_1-4-2011.DS17706.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD3_Primary_
Cells.CB6_1-4-
2011+ | Blood | Hotspots | CD3 | CB6_1-4-
2011+ | GSM701526 |
UW.CD3_Primary_Cells.ChromatinAccessibility.
RO_01679.DS17198.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD3_Primary_
Cells.RO_01679+ | Blood | Hotspots | CD3 | RO_01679+ | GSM665837 |
UW.CD3_Primary_Cells.ChromatinAccessibility.
RO_01701.DS17534.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD3_Primary_
Cells.RO_01701+ | Blood | Hotspots | CD3 | RO_01701+ | GSM701516 |
UW.CD4_Primary_Cells.ChromatinAccessibility.
RO_01549.DS15947.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD4_Primary_
Cells.RO_01549+ | Blood | Hotspots | CD4 | RO_01549+ | GSM665812 |
UW.CD4_Primary_Cells.ChromatinAccessibility.
RO_01679.DS17212.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD4_Primary_
Cells.RO_01679+ | Blood | Hotspots | CD4 | RO_01679+ | GSM665839 |
UW.CD4_Primary_Cells.ChromatinAccessibility.
RO_01689.DS17329.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD4_Primary_
Cells.RO_01689+ | Blood | Hotspots | CD4 | RO_01689+ | GSM817166 |
UW.CD4_Primary_Cells.ChromatinAccessibility.
RO_01701.DS17881.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD4_Primary_
Cells.RO_01701+ | Blood | Hotspots | CD4 | RO_01701+ | GSM701539 |
UW.CD56_Primary_Cells.ChromatinAccessibility.
RO_01679.DS17189.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD56_Primary_
Cells.RO_01679+ | Blood | Hotspots | CD56 | RO_01679+ | GSM665836 |
UW.CD56_Primary_Cells.ChromatinAccessibility.
RO_01701.DS17443.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD56_Primary_
Cells.RO_01701+ | Blood | Hotspots | CD56 | RO_01701+ | GSM701508 |
UW.CD8_Primary_Cells.ChromatinAccessibility.
RO_01549.DS16012.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD8_Primary_
Cells.RO_01549+ | Blood | Hotspots | CD8 | RO_01549+ | GSM665813 |
UW.CD8_Primary_Cells.ChromatinAccessibility.
RO_01679.DS17203.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD8_Primary_
Cells.RO_01679+ | Blood | Hotspots | CD8 | RO_01679+ | GSM665838 |
UW.CD8_Primary_Cells.ChromatinAccessibility.
RO_01689.DS17332.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD8_Primary_
Cells.RO_01689+ | Blood | Hotspots | CD8 | RO_01689+ | GSM701499 |
UW.CD8_Primary_Cells.ChromatinAccessibility.
RO_01701.DS17885.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | CD8_Primary_
Cells.RO_01701+ | Blood | Hotspots | CD8 | RO_01701+ | GSM701540 |
UW.Fetal_Adrenal_Gland.ChromatinAccessibility.
H-22662.DS12528.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Adrenal_
Gland.H-22662 | Fetal Adrenal | Hotspots | Fetal_Adrenal_
Gland | H-22662 | GSM530653 |
UW.Fetal_Adrenal_Gland.ChromatinAccessibility.
H-23769.DS17319.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Adrenal_
Gland.H-23769 | Fetal Adrenal | Hotspots | Fetal_Adrenal_
Gland | H-23769 | GSM817165 |
UW.Fetal_Adrenal_Gland.ChromatinAccessibility.
H-24409.DS20343.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Adrenal_
Gland.H-24409 | Fetal Adrenal | Hotspots | Fetal_Adrenal_
Gland | H-24409 | GSM878658 |
UW.Fetal_Brain.ChromatinAccessibility.H-22510.
DS11872.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Brain.
H-22510 | Fetal Brain | Hotspots | Fetal_Brain | H-22510 | GSM530651 |
UW.Fetal_Brain.ChromatinAccessibility.H-22510.
DS11877.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Brain.
H-22510 | Fetal Brain | Hotspots | Fetal_Brain | H-22510 | GSM595913 |
UW.Fetal_Brain.ChromatinAccessibility.H-22911.
DS14464.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Brain.
H-22911 | Fetal Brain | Hotspots | Fetal_Brain | H-22911 | GSM595920 |
UW.Fetal_Brain.ChromatinAccessibility.H-23266.
DS14717.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Brain.
H-23266 | Fetal Brain | Hotspots | Fetal_Brain | H-23266 | GSM595922 |
UW.Fetal_Brain.ChromatinAccessibility.H-23266.
DS14718.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Brain.
H-23266 | Fetal Brain | Hotspots | Fetal_Brain | H-23266 | GSM595923 |
UW.Fetal_Brain.ChromatinAccessibility.H-23284.
DS14803.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Brain.
H-23284 | Fetal Brain | Hotspots | Fetal_Brain | H-23284 | GSM595926 |
UW.Fetal_Brain.ChromatinAccessibility.H-23284.
DS14815.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Brain.
H-23284 | Fetal Brain | Hotspots | Fetal_Brain | H-23284 | GSM595928 |
UW.Fetal_Brain.ChromatinAccessibility.H-23399.
DS15453.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Brain.
H-23399 | Fetal Brain | Hotspots | Fetal_Brain | H-23399 | GSM665804 |
UW.Fetal_Brain.ChromatinAccessibility.H-23548.
DS16302.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Brain.
H-23548 | Fetal Brain | Hotspots | Fetal_Brain | H-23548 | GSM665819 |
UW.Fetal_Brain.ChromatinAccessibility.H-24279.
DS20221.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Brain.
H-24279 | Fetal Brain | Hotspots | Fetal_Brain | H-24279 | GSM878650 |
UW.Fetal_Brain.ChromatinAccessibility.H-24297.
DS20226.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Brain.
H-24297 | Fetal Brain | Hotspots | Fetal_Brain | H-24297 | GSM878651 |
UW.Fetal_Brain.ChromatinAccessibility.H-24381.
DS20231.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Brain.
H-24381 | Fetal Brain | Hotspots | Fetal_Brain | H-24381 | GSM878652 |
UW.Fetal_Heart.ChromatinAccessibility.H-22662.
DS12531.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Heart.
H-22662 | Fetal Heart | Hotspots | Fetal_Heart | H-22662 | GSM530654 |
UW.Fetal_Heart.ChromatinAccessibility.H-22727.
DS12810.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Heart.
H-22727 | Fetal Heart | Hotspots | Fetal_Heart | H-22727 | GSM530661 |
UW.Fetal_Heart.ChromatinAccessibility.H-23468.
DS15839.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Heart.
H-23468 | Fetal Heart | Hotspots | Fetal_Heart | H-23468 | GSM665811 |
UW.Fetal_Heart.ChromatinAccessibility.H-23500.
DS16018.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Heart.
H-23500 | Fetal Heart | Hotspots | Fetal_Heart | H-23500 | GSM665814 |
UW.Fetal_Heart.ChromatinAccessibility.H-23524.
DS16146.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Heart.
H-23524 | Fetal Heart | Hotspots | Fetal_Heart | H-23524 | GSM665817 |
UW.Fetal_Heart.ChromatinAccessibility.H-23589.
DS16500.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Heart.
H-23589 | Fetal Heart | Hotspots | Fetal_Heart | H-23589 | GSM665824 |
UW.Fetal_Heart.ChromatinAccessibility.H-23604
.DS16582.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Heart.
H-23604 | Fetal Heart | Hotspots | Fetal_Heart | H-23604 | GSM665830 |
UW.Fetal_Heart.ChromatinAccessibility.H-23617.
DS16621.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Heart.
H-23617 | Fetal Heart | Hotspots | Fetal_Heart | H-23617 | GSM665831 |
UW.Fetal_Heart.ChromatinAccessibility.H-23663.
DS16819.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Heart.
H-23663 | Fetal Heart | Hotspots | Fetal_Heart | H-23663 | GSM774203 |
UW.Fetal_Heart.ChromatinAccessibility.H-23744.
DS19431.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Heart.
H-23744 | Fetal Heart | Hotspots | Fetal_Heart | H-23744 | GSM817220 |
UW.Fetal_Heart.ChromatinAccessibility.H-24042.
DS19427.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Heart.
H-24042 | Fetal Heart | Hotspots | Fetal_Heart | H-24042 | GSM878630 |
UW.Fetal_Intestine_Large.ChromatinAccessibility.H-
23500.DS16027.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Large.H-23500 | Fetal Intestine,
Large | Hotspots | Fetal_Intestine_
Large | H-23500 | GSM665815 |
UW.Fetal_Intestine_Large.ChromatinAccessibility.H-
23524.DS16164.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Large.H-23524 | Fetal Intestine,
Large | Hotspots | Fetal_Intestine_
Large | H-23524 | GSM665818 |
UW.Fetal_Intestine_Large.ChromatinAccessibility.H-
23604.DS16563.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Large.H-23604 | Fetal Intestine,
Large | Hotspots | Fetal_Intestine_
Large | H-23604 | GSM665826 |
UW.Fetal_Intestine_Large.ChromatinAccessibility.H-
23744.DS17094.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Large.H-23744 | Fetal Intestine,
Large | Hotspots | Fetal_Intestine_
Large | H-23744 | GSM817162 |
UW.Fetal_Intestine_Large.ChromatinAccessibility.H-
23758.DS17157.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Large.H-23758 | Fetal Intestine,
Large | Hotspots | Fetal_Intestine_
Large | H-23758 | GSM701490 |
UW.Fetal_Intestine_Large.ChromatinAccessibility.H-
23769.DS17313.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Large.H-23769 | Fetal Intestine,
Large | Hotspots | Fetal_Intestine_
Large | H-23769 | GSM701495 |
UW.Fetal_Intestine_Large.ChromatinAccessibility.H-
23808.DS17422.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Large.H-23808 | Fetal Intestine,
Large | Hotspots | Fetal_Intestine_
Large | H-23808 | GSM774213 |
UW.Fetal_Intestine_Large.ChromatinAccessibility.H-
23833.DS17462.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Large.H-23833 | Fetal Intestine,
Large | Hotspots | Fetal_Intestine_
Large | H-23833 | GSM774214 |
UW.Fetal_Intestine_Large.ChromatinAccessibility.H-
23855.DS17502.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Large.H-23855 | Fetal Intestine,
Large | Hotspots | Fetal_Intestine_
Large | H-23855 | GSM701514 |
UW.Fetal_Intestine_Large.ChromatinAccessibility.H-
23887.DS17647.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Large.H-23887 | Fetal Intestine,
Large | Hotspots | Fetal_Intestine_
Large | H-23887 | GSM774217 |
UW.Fetal_Intestine_Large.ChromatinAccessibility.H-
23914.DS17748.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Large.H-23914 | Fetal Intestine,
Large | Hotspots | Fetal_Intestine_
Large | H-23914 | GSM774220 |
UW.Fetal_Intestine_Large.ChromatinAccessibility.H-
23941.DS17785.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Large.H-23941 | Fetal Intestine,
Large | Hotspots | Fetal_Intestine_
Large | H-23941 | GSM701531 |
UW.Fetal_Intestine_Large.ChromatinAccessibility.H-
23964.DS17841.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Large.H-23964 | Fetal Intestine,
Large | Hotspots | Fetal_Intestine_
Large | H-23964 | GSM774228 |
UW.Fetal_Intestine_Large.ChromatinAccessibility.H-
24005.DS17990.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Large.H-24005 | Fetal Intestine,
Large | Hotspots | Fetal_Intestine_
Large | H-24005 | GSM774233 |
UW.Fetal_Intestine_Large.ChromatinAccessibility.H-
24111.DS18499.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Large.H-24111 | Fetal Intestine,
Large | Hotspots | Fetal_Intestine_
Large | H-24111 | GSM817188 |
UW.Fetal_Intestine_Small.ChromatinAccessibility.H-
23604.DS16559.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Small.H-23604 | Fetal Intestine,
Small | Hotspots | Fetal_Intestine_
Small | H-23604 | GSM665825 |
UW.Fetal_Intestine_Small.ChromatinAccessibility.H-
23640.DS16712.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Small.H-23640 | Fetal Intestine,
Small | Hotspots | Fetal_Intestine_
Small | H-23640 | GSM701487 |
UW.Fetal_Intestine_Small.ChromatinAccessibility.H-
23663.DS16822.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Small.H-23663 | Fetal Intestine,
Small | Hotspots | Fetal_Intestine_
Small | H-23663 | GSM665835 |
UW.Fetal_Intestine_Small.ChromatinAccessibility.
H-23724.DS16975.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Small.H-23724 | Fetal Intestine,
Small | Hotspots | Fetal_Intestine_
Small | H-23724 | GSM817161 |
UW.Fetal_Intestine_Small.ChromatinAccessibility.
H-23744.DS17092.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Small.H-23744 | Fetal Intestine,
Small | Hotspots | Fetal_Intestine_
Small | H-23744 | GSM774205 |
UW.Fetal_Intestine_Small.ChromatinAccessibility.
H-23758.DS17150.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Small.H-23758 | Fetal Intestine,
Small | Hotspots | Fetal_Intestine_
Small | H-23758 | GSM774210 |
UW.Fetal_Intestine_Small.ChromatinAccessibility.
H-23769.DS17317.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Small.H-23769 | Fetal Intestine,
Small | Hotspots | Fetal_Intestine_
Small | H-23769 | GSM701496 |
UW.Fetal_Intestine_Small.ChromatinAccessibility.
H-23808.DS17425.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Small.H-23808 | Fetal Intestine,
Small | Hotspots | Fetal_Intestine_
Small | H-23808 | GSM701504 |
UW.Fetal_Intestine_Small.ChromatinAccessibility.
H-23864.DS17844.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Small.H-23864 | Fetal Intestine,
Small | Hotspots | Fetal_Intestine_
Small | H-23864 | GSM774229 |
UW.Fetal_Intestine_Small.ChromatinAccessibility.
H-23887.DS17643.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Small.H-23887 | Fetal Intestine,
Small | Hotspots | Fetal_Intestine_
Small | H-23887 | GSM774216 |
UW.Fetal_Intestine_Small.ChromatinAccessibility.
H-23914.DS17763.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Small.H-23914 | Fetal Intestine,
Small | Hotspots | Fetal_Intestine_
Small | H-23914 | GSM701530 |
UW.Fetal_Intestine_Small.ChromatinAccessibility.
H-23941.DS17808.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Small.H-23941 | Fetal Intestine,
Small | Hotspots | Fetal_Intestine_
Small | H-23941 | GSM774225 |
UW.Fetal_Intestine_Small.ChromatinAccessibility.
H-24111.DS18495.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Intestine_
Small.H-24111 | Fetal Intestine,
Small | Hotspots | Fetal_Intestine_
Small | H-24111 | GSM817187 |
UW.Fetal_Kidney.ChromatinAccessibility.H-22337.
DS10986.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Kidney.
H-22337 | Fetal Kidney | Hotspots | Fetal_Kidney | H-22337 | GSM493385 |
UW.Fetal_Kidney.ChromatinAccessibility.H-22676.
DS12635.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Kidney.
H-22676 | Fetal Kidney | Hotspots | Fetal_Kidney | H-22676 | GSM530655 |
UW.Fetal_Kidney.ChromatinAccessibility.H-23524.
DS16139.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Kidney.
H-23524 | Fetal Kidney | Hotspots | Fetal_Kidney | H-23524 | GSM665816 |
UW.Fetal_Kidney.ChromatinAccessibility.H-23663.
DS16837.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Kidney.
H-23663 | Fetal Kidney | Hotspots | Fetal_Kidney | H-23663 | GSM817159 |
UW.Fetal_Kidney.ChromatinAccessibility.H-23855.
DS17522.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Kidney.
H-23855 | Fetal Kidney | Hotspots | Fetal_Kidney | H-23855 | GSM701515 |
UW.Fetal_Kidney.ChromatinAccessibility.H-23914.
DS17753.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Kidney.
H-23914 | Fetal Kidney | Hotspots | Fetal_Kidney | H-23914 | GSM774221 |
UW.Fetal_Kidney.ChromatinAccessibility.H-24507.
DS20564.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Kidney.
H-24507 | Fetal Kidney | Hotspots | Fetal_Kidney | H-24507 | GSM878666 |
UW.Fetal_Kidney_Left.ChromatinAccessibility.H-23589.
DS16446.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Kidney_
Left.H-23589 | Fetal Kidney | Hotspots | Fetal_Kidney_Left | H-23589 | GSM665822 |
UW.Fetal_Kidney_Left.ChromatinAccessibility.H-23604.
DS16579.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Kidney_
Left.H-23604 | Fetal Kidney | Hotspots | Fetal_Kidney_Left | H-23604 | GSM665829 |
UW.Fetal_Kidney_Left.ChromatinAccessibility.H-23640.
DS16805.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Kidney_
Left.H-23640 | Fetal Kidney | Hotspots | Fetal_Kidney_Left | H-23640 | GSM665834 |
UW.Fetal_Kidney_Left.ChromatinAccessibility.H-23758.
DS17140.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Kidney_
Left.H-23758 | Fetal Kidney | Hotspots | Fetal_Kidney_Left | H-23758 | GSM774209 |
UW.Fetal_Kidney_Left.ChromatinAccessibility.H-24089.
DS18466.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Kidney_
Left.H-24089 | Fetal Kidney | Hotspots | Fetal_Kidney_Left | H-24089 | GSM817181 |
UW.Fetal_Kidney_Right.ChromatinAccessibility.H-23435.
DS15651.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Kidney_
Right.H-23435 | Fetal Kidney | Hotspots | Fetal_Kidney_
Right | H-23435 | GSM665810 |
UW.Fetal_Kidney_Right.ChromatinAccessibility.H-23589.
DS16441.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Kidney_
Right.H-23589 | Fetal Kidney | Hotspots | Fetal_Kidney_
Right | H-23589 | GSM665821 |
UW.Fetal_Kidney_Right.ChromatinAccessibility.H-23640.
DS16801.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Kidney_
Right.H-23640 | Fetal Kidney | Hotspots | Fetal_Kidney_
Right | H-23640 | GSM665833 |
UW.Fetal_Kidney_Right.ChromatinAccessibility.H-23758.
DS17144.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Kidney_
Right.H-23758 | Fetal Kidney | Hotspots | Fetal_Kidney_
Right | H-23758 | GSM817163 |
UW.Fetal_Kidney_Right.ChromatinAccessibility.H-24089.
DS18463.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Kidney_
Right.H-24089 | Fetal Kidney | Hotspots | Fetal_Kidney_
Right | H-24089 | GSM817180 |
UW.Fetal_Lung.ChromatinAccessibility.H-22676.
DS12646.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung.
H-22676 | Fetal Lung | Hotspots | Fetal_Lung | H-22676 | GSM530656 |
UW.Fetal_Lung.ChromatinAccessibility.H-22727.
DS12817.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung.
H-22727 | Fetal Lung | Hotspots | Fetal_Lung | H-22727 | GSM530662 |
UW.Fetal_Lung.ChromatinAccessibility.H-22934B.
DS13507.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung.
H-22934B | Fetal Lung | Hotspots | Fetal_Lung | H-22934B | GSM595915 |
UW.Fetal_Lung.ChromatinAccessibility.H-23090.
DS13985.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung.
H-23090 | Fetal Lung | Hotspots | Fetal_Lung | H-23090 | GSM595916 |
UW.Fetal_Lung.ChromatinAccessibility.H-23247.
DS14666.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung.
H-23247 | Fetal Lung | Hotspots | Fetal_Lung | H-23247 | GSM595921 |
UW.Fetal_Lung.ChromatinAccessibility.H-23266.
DS14724.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung.
H-23266 | Fetal Lung | Hotspots | Fetal_Lung | H-23266 | GSM595924 |
UW.Fetal_Lung.ChromatinAccessibility.H-23266.
DS14751.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung.
H-23266 | Fetal Lung | Hotspots | Fetal_Lung | H-23266 | GSM595925 |
UW.Fetal_Lung.ChromatinAccessibility.H-23284.
DS14809.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung.
H-23284 | Fetal Lung | Hotspots | Fetal_Lung | H-23284 | GSM595927 |
UW.Fetal_Lung.ChromatinAccessibility.H-23284.
DS14820.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung.
H-23284 | Fetal Lung | Hotspots | Fetal_Lung | H-23284 | GSM595929 |
UW.Fetal_Lung.ChromatinAccessibility.H-23365.
DS15227.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung.
H-23365 | Fetal Lung | Hotspots | Fetal_Lung | H-23365 | GSM595930 |
UW.Fetal_Lung.ChromatinAccessibility.H-23399.
DS15461.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung.
H-23399 | Fetal Lung | Hotspots | Fetal_Lung | H-23399 | GSM665805 |
UW.Fetal_Lung.ChromatinAccessibility.H-23419.
DS15573.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung.
H-23419 | Fetal Lung | Hotspots | Fetal_Lung | H-23419 | GSM665806 |
UW.Fetal_Lung_Left.ChromatinAccessibility.H-23435.
DS15637.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_Left.
H-23435 | Fetal Lung | Hotspots | Fetal_Lung_Left | H-23435 | GSM665808 |
UW.Fetal_Lung_Left.ChromatinAccessibility.H-23604.
DS16570.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_Left.
H-23604 | Fetal Lung | Hotspots | Fetal_Lung_Left | H-23604 | GSM665828 |
UW.Fetal_Lung_Left.ChromatinAccessibility.H-23640.
DS18170.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_Left.
H-23640 | Fetal Lung | Hotspots | Fetal_Lung_Left | H-23640 | GSM774237 |
UW.Fetal_Lung_Left.ChromatinAccessibility.H-23744.
DS17105.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_Left.
H-23744 | Fetal Lung | Hotspots | Fetal_Lung_Left | H-23744 | GSM774207 |
UW.Fetal_Lung_Left.ChromatinAccessibility.H-23758.
DS17154.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_Left.
H-23758 | Fetal Lung | Hotspots | Fetal_Lung_Left | H-23758 | GSM774211 |
UW.Fetal_Lung_Left.ChromatinAccessibility.H-23833.
DS17464.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_Left.
H-23833 | Fetal Lung | Hotspots | Fetal_Lung_Left | H-23833 | GSM701512 |
UW.Fetal_Lung_Left.ChromatinAccessibility.H-23887.
DS17674.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_Left.
H-23887 | Fetal Lung | Hotspots | Fetal_Lung_Left | H-23887 | GSM701524 |
UW.Fetal_Lung_Left.ChromatinAccessibility.H-23914.
DS17739.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_Left.
H-23914 | Fetal Lung | Hotspots | Fetal_Lung_Left | H-23914 | GSM701527 |
UW.Fetal_Lung_Left.ChromatinAccessibility.H-23964.
DS17835.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_Left.
H-23964 | Fetal Lung | Hotspots | Fetal_Lung_Left | H-23964 | GSM701534 |
UW.Fetal_Lung_Left.ChromatinAccessibility.H-24005.
DS17959.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_Left.
H-24005 | Fetal Lung | Hotspots | Fetal_Lung_Left | H-24005 | GSM817168 |
UW.Fetal_Lung_Left.ChromatinAccessibility.H-24089.
DS18421.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_Left.
H-24089 | Fetal Lung | Hotspots | Fetal_Lung_Left | H-24089 | GSM817175 |
UW.Fetal_Lung_Left.ChromatinAccessibility.H-24111.
DS18487.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_Left.
H-24111 | Fetal Lung | Hotspots | Fetal_Lung_Left | H-24111 | GSM817185 |
UW.Fetal_Lung_Right.ChromatinAccessibility.H-23435.
DS15632.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_
Right.H-23435 | Fetal Lung | Hotspots | Fetal_Lung_Right | H-23435 | GSM665807 |
UW.Fetal_Lung_Right.ChromatinAccessibility.H-23604.
DS16566.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_
Right.H-23604 | Fetal Lung | Hotspots | Fetal_Lung_Right | H-23604 | GSM665827 |
UW.Fetal_Lung_Right.ChromatinAccessibility.H-23640.
DS16790.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_
Right.H-23640 | Fetal Lung | Hotspots | Fetal_Lung_Right | H-23640 | GSM665832 |
UW.Fetal_Lung_Right.ChromatinAccessibility.H-23744.
DS17101.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_
Right.H-23744 | Fetal Lung | Hotspots | Fetal_Lung_Right | H-23744 | GSM774206 |
UW.Fetal_Lung_Right.ChromatinAccessibility.H-23758.
DS17162.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_
Right.H-23758 | Fetal Lung | Hotspots | Fetal_Lung_Right | H-23758 | GSM817164 |
UW.Fetal_Lung_Right.ChromatinAccessibility.H-23887.
DS17670.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_
Right.H-23887 | Fetal Lung | Hotspots | Fetal_Lung_Right | H-23887 | GSM701523 |
UW.Fetal_Lung_Right.ChromatinAccessibility.H-23964.
DS17831.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_
Right.H-23964 | Fetal Lung | Hotspots | Fetal_Lung_Right | H-23964 | GSM774227 |
UW.Fetal_Lung_Right.ChromatinAccessibility.H-24005.
DS17954.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_
Right.H-24005 | Fetal Lung | Hotspots | Fetal_Lung_Right | H-24005 | GSM774231 |
UW.Fetal_Lung_Right.ChromatinAccessibility.H-24089.
DS18418.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_
Right.H-24089 | Fetal Lung | Hotspots | Fetal_Lung_Right | H-24089 | GSM817174 |
UW.Fetal_Lung_Right.ChromatinAccessibility.H-24111.
DS18492.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Lung_
Right.H-24111 | Fetal Lung | Hotspots | Fetal_Lung_Right | H-24111 | GSM817186 |
UW.Fetal_Muscle_Arm.ChromatinAccessibility.H-23808.
DS17432.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Arm.H-23808 | Fetal Muscle | Hotspots | Fetal_Muscle_
Arm | H-23808 | GSM701506 |
UW.Fetal_Muscle_Arm.ChromatinAccessibility.H-23914.
DS17765.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Arm.H-23914 | Fetal Muscle | Hotspots | Fetal_Muscle_
Arm | H-23914 | GSM774223 |
UW.Fetal_Muscle_Arm.ChromatinAccessibility.H-23941.
DS17825.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Arm.H-23941 | Fetal Muscle | Hotspots | Fetal_Muscle_
Arm | H-23941 | GSM774226 |
UW.Fetal_Muscle_Arm.ChromatinAccessibility.H-23964.
DS17848.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Arm.H-23964 | Fetal Muscle | Hotspots | Fetal_Muscle_
Arm | H-23964 | GSM701535 |
UW.Fetal_Muscle_Arm.ChromatinAccessibility.H-24005.
DS18080.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Arm.H-24005 | Fetal Muscle | Hotspots | Fetal_Muscle_
Arm | H-24005 | GSM774234 |
UW.Fetal_Muscle_Arm.ChromatinAccessibility.H-24042.
DS18176.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Arm.H-24042 | Fetal Muscle | Hotspots | Fetal_Muscle_
Arm | H-24042 | GSM774239 |
UW.Fetal_Muscle_Arm.ChromatinAccessibility.H-24078.
DS18379.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Arm.H-24078 | Fetal Muscle | Hotspots | Fetal_Muscle_
Arm | H-24078 | GSM878610 |
UW.Fetal_Muscle_Arm.ChromatinAccessibility.H-24089.
DS18452.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Arm.H-24089 | Fetal Muscle | Hotspots | Fetal_Muscle_
Arm | H-24089 | GSM817178 |
UW.Fetal_Muscle_Arm.ChromatinAccessibility.H-24111.
DS18473.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Arm.H-24111 | Fetal Muscle | Hotspots | Fetal_Muscle_
Arm | H-24111 | GSM817184 |
UW.Fetal_Muscle_Arm.ChromatinAccessibility.H-24125.
DS18559.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Arm.H-24125 | Fetal Muscle | Hotspots | Fetal_Muscle_
Arm | H-24125 | GSM817191 |
UW.Fetal_Muscle_Arm.ChromatinAccessibility.H-24143.
DS18992.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Arm.H-24143 | Fetal Muscle | Hotspots | Fetal_Muscle_
Arm | H-24143 | GSM878618 |
UW.Fetal_Muscle_Arm.ChromatinAccessibility.H-24198.
DS19051.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Arm.H-24198 | Fetal Muscle | Hotspots | Fetal_Muscle_
Arm | H-24198 | GSM878619 |
UW.Fetal_Muscle_Arm.ChromatinAccessibility.H-24218.
DS19053.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Arm.H-24218 | Fetal Muscle | Hotspots | Fetal_Muscle_
Arm | H-24218 | GSM878620 |
UW.Fetal_Muscle_Arm.ChromatinAccessibility.H-24244.
DS19270.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Arm.H-24244 | Fetal Muscle | Hotspots | Fetal_Muscle_
Arm | H-24244 | GSM878625 |
UW.Fetal_Muscle_Arm.ChromatinAccessibility.H-24259.
DS19295.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Arm.H-24259 | Fetal Muscle | Hotspots | Fetal_Muscle_
Arm | H-24259 | GSM817214 |
UW.Fetal_Muscle_Arm.ChromatinAccessibility.H-24272.
DS19380.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Arm.H-24272 | Fetal Muscle | Hotspots | Fetal_Muscle_
Arm | H-24272 | GSM817216 |
UW.Fetal_Muscle_Arm.ChromatinAccessibility.H-24297.
DS19646.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Arm.H-24297 | Fetal Muscle | Hotspots | Fetal_Muscle_
Arm | H-24297 | GSM878638 |
UW.Fetal_Muscle_Back.ChromatinAccessibility.H-23914.
DS17767.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Back.H-23914 | Fetal Muscle | Hotspots | Fetal_Muscle_
Back | H-23914 | GSM774224 |
UW.Fetal_Muscle_Back.ChromatinAccessibility.H-23964.
DS17850.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Back.H-23964 | Fetal Muscle | Hotspots | Fetal_Muscle_
Back | H-23964 | GSM701536 |
UW.Fetal_Muscle_Back.ChromatinAccessibility.H-24005.
DS18083.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Back.H-24005 | Fetal Muscle | Hotspots | Fetal_Muscle_
Back | H-24005 | GSM774235 |
UW.Fetal_Muscle_Back.ChromatinAccessibility.H-24078.
DS18377.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Back.H-24078 | Fetal Muscle | Hotspots | Fetal_Muscle_
Back | H-24078 | GSM817171 |
UW.Fetal_Muscle_Back.ChromatinAccessibility.H-24089.
DS18454.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Back.H-24089 | Fetal Muscle | Hotspots | Fetal_Muscle_
Back | H-24089 | GSM878611 |
UW.Fetal_Muscle_Back.ChromatinAccessibility.H-24111.
DS18468.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Back.H-24111 | Fetal Muscle | Hotspots | Fetal_Muscle_
Back | H-24111 | GSM817182 |
UW.Fetal_Muscle_Back.ChromatinAccessibility.H-24143.
DS18842.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Back.H-24143 | Fetal Muscle | Hotspots | Fetal_Muscle_
Back | H-24143 | GSM817200 |
UW.Fetal_Muscle_Back.ChromatinAccessibility.H-24218.
DS19117.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Back.H-24218 | Fetal Muscle | Hotspots | Fetal_Muscle_
Back | H-24218 | GSM817207 |
UW.Fetal_Muscle_Back.ChromatinAccessibility.H-24244.
DS19283.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Back.H-24244 | Fetal Muscle | Hotspots | Fetal_Muscle_
Back | H-24244 | GSM817212 |
UW.Fetal_Muscle_Back.ChromatinAccessibility.H-24272.
DS19384.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Back.H-24272 | Fetal Muscle | Hotspots | Fetal_Muscle_
Back | H-24272 | GSM817217 |
UW.Fetal_Muscle_Back.ChromatinAccessibility.H-24279.
DS19441.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Back.H-24279 | Fetal Muscle | Hotspots | Fetal_Muscle_
Back | H-24279 | GSM878632 |
UW.Fetal_Muscle_Back.ChromatinAccessibility.H-24297.
DS19648.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Back.H-24297 | Fetal Muscle | Hotspots | Fetal_Muscle_
Back | H-24297 | GSM878639 |
UW.Fetal_Muscle_Back.ChromatinAccessibility.H-24409.
DS20244.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Back.H-24409 | Fetal Muscle | Hotspots | Fetal_Muscle_
Back | H-24409 | GSM878655 |
UW.Fetal_Muscle_Leg.ChromatinAccessibility.H-23808.
DS17429.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Leg.H-23808 | Fetal Muscle | Hotspots | Fetal_Muscle_
Leg | H-23808 | GSM701505 |
UW.Fetal_Muscle_Leg.ChromatinAccessibility.H-24078.
DS18386.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Leg.H-24078 | Fetal Muscle | Hotspots | Fetal_Muscle_
Leg | H-24078 | GSM774242 |
UW.Fetal_Muscle_Leg.ChromatinAccessibility.H-24089.
DS18456.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Leg.H-24089 | Fetal Muscle | Hotspots | Fetal_Muscle_
Leg | H-24089 | GSM817179 |
UW.Fetal_Muscle_Leg.ChromatinAccessibility.H-24111.
DS18471.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Leg.H-24111 | Fetal Muscle | Hotspots | Fetal_Muscle_
Leg | H-24111 | GSM817183 |
UW.Fetal_Muscle_Leg.ChromatinAccessibility.H-24143.
DS18844.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Leg.H-24143 | Fetal Muscle | Hotspots | Fetal_Muscle_
Leg | H-24143 | GSM817201 |
UW.Fetal_Muscle_Leg.ChromatinAccessibility.H-24198.
DS19158.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Leg.H-24198 | Fetal Muscle | Hotspots | Fetal_Muscle_
Leg | H-24198 | GSM878622 |
UW.Fetal_Muscle_Leg.ChromatinAccessibility.H-24218.
DS19115.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Leg.H-24218 | Fetal Muscle | Hotspots | Fetal_Muscle_
Leg | H-24218 | GSM817206 |
UW.Fetal_Muscle_Leg.ChromatinAccessibility.H-24244.
DS19272.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Leg.H-24244 | Fetal Muscle | Hotspots | Fetal_Muscle_
Leg | H-24244 | GSM878626 |
UW.Fetal_Muscle_Leg.ChromatinAccessibility.H-24259.
DS19291.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Leg.H-24259 | Fetal Muscle | Hotspots | Fetal_Muscle_
Leg | H-24259 | GSM817213 |
UW.Fetal_Muscle_Leg.ChromatinAccessibility.H-24279.
DS19436.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Leg.H-24279 | Fetal Muscle | Hotspots | Fetal_Muscle_
Leg | H-24279 | GSM878631 |
UW.Fetal_Muscle_Leg.ChromatinAccessibility.H-24297.
DS19643.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Leg.H-24297 | Fetal Muscle | Hotspots | Fetal_Muscle_
Leg | H-24297 | GSM878637 |
UW.Fetal_Muscle_Leg.ChromatinAccessibility.H-24409.
DS20239.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Leg.H-24409 | Fetal Muscle | Hotspots | Fetal_Muscle_
Leg | H-24409 | GSM878653 |
UW.Fetal_Muscle_Lower_Limb_Skeletal.
ChromatinAccessibility.H-24042.DS18174.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Lower_Limb_
Skeletal.H-24042 | Fetal Muscle | Hotspots | Fetal_Muscle_
Lower_Limb_
Skeletal | H-24042 | GSM774238 |
UW.Fetal_Muscle_Trunk.ChromatinAccessibility.H-23941.
DS17827.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Trunk.H-23941 | Fetal Muscle | Hotspots | Fetal_Muscle_
Trunk | H-23941 | GSM701533 |
UW.Fetal_Muscle_Trunk.ChromatinAccessibility.H-24409.
DS20242.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Trunk.H-24409 | Fetal Muscle | Hotspots | Fetal_Muscle_
Trunk | H-24409 | GSM878654 |
UW.Fetal_Muscle_Trunk.ChromatinAccessibility.H-24507.
DS20544.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Trunk.H-24507 | Fetal Muscle | Hotspots | Fetal_Muscle_
Trunk | H-24507 | GSM878664 |
UW.Fetal_Muscle_Upper_Limb_Skeletal.
ChromatinAccessibility.H-23887.DS17661.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Upper_Limb_
Skeletal.H-23887 | Fetal Muscle | Hotspots | Fetal_Muscle_
Upper_Limb_
Skeletal | H-23887 | GSM701522 |
UW.Fetal_Muscle_Upper_Trunk.ChromatinAccessibility.
H-23887.DS17664.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Muscle_
Upper_Trunk.
H-23887 | Fetal Muscle | Hotspots | Fetal_Muscle_
Upper_Trunk | H-23887 | GSM774218 |
UW.Fetal_Placenta.ChromatinAccessibility.H-23887.
DS17639.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Placenta.
H-23887 | Fetal Placenta | Hotspots | Fetal_Placenta | H-23887 | GSM774215 |
UW.Fetal_Placenta.ChromatinAccessibility.H-23914.
DS17744.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Placenta.
H-23914 | Fetal Placenta | Hotspots | Fetal_Placenta | H-23914 | GSM774219 |
UW.Fetal_Placenta.ChromatinAccessibility.H-24272.
DS19391.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Placenta.
H-24272 | Fetal Placenta | Hotspots | Fetal_Placenta | H-24272 | GSM817219 |
UW.Fetal_Placenta.ChromatinAccessibility.H-24409.
DS20346.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Placenta.
H-24409 | Fetal Placenta | Hotspots | Fetal_Placenta | H-24409 | GSM878659 |
UW.Fetal_Renal_Cortex.ChromatinAccessibility.H-23769.
DS17307.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Cortex.H-23769 | Fetal Renal
Cortex | Hotspots | Fetal_Renal_
Cortex | H-23769 | GSM701494 |
UW.Fetal_Renal_Cortex.ChromatinAccessibility.H-23790.
DS17387.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Cortex.H-23790 | Fetal Renal
Cortex | Hotspots | Fetal_Renal_
Cortex | H-23790 | GSM701502 |
UW.Fetal_Renal_Cortex.ChromatinAccessibility.H-23833.
DS17455.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Cortex.H-23833 | Fetal Renal
Cortex | Hotspots | Fetal_Renal_
Cortex | H-23833 | GSM701511 |
UW.Fetal_Renal_Cortex.ChromatinAccessibility.H-23914.
DS17756.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Cortex.H-23914 | Fetal Renal
Cortex | Hotspots | Fetal_Renal_
Cortex | H-23914 | GSM701529 |
UW.Fetal_Renal_Cortex.ChromatinAccessibility.H-23941.
DS17804.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Cortex.H-23941 | Fetal Renal
Cortex | Hotspots | Fetal_Renal_
Cortex | H-23941 | GSM701532 |
UW.Fetal_Renal_Cortex.ChromatinAccessibility.H-24078.
DS18428.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Cortex.H-24078 | Fetal Renal
Cortex | Hotspots | Fetal_Renal_
Cortex | H-24078 | GSM817176 |
UW.Fetal_Renal_Cortex.ChromatinAccessibility.H-24259.
DS19388.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Cortex.H-24259 | Fetal Renal
Cortex | Hotspots | Fetal_Renal_
Cortex | H-24259 | GSM878629 |
UW.Fetal_Renal_Cortex.ChromatinAccessibility.H-24493.
DS20568.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Cortex.H-24493 | Fetal Renal
Cortex | Hotspots | Fetal_Renal_
Cortex | H-24493 | GSM878667 |
UW.Fetal_Renal_Cortex_Left.ChromatinAccessibility.
H-23871.DS17550.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Cortex_Left.
H-23871 | Fetal Renal
Cortex | Hotspots | Fetal_Renal_
Cortex_Left | H-23871 | GSM701519 |
UW.Fetal_Renal_Cortex_Left.ChromatinAccessibility.
H-24111.DS18542.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Cortex_Left.
H-24111 | Fetal Renal
Cortex | Hotspots | Fetal_Renal_
Cortex_Left | H-24111 | GSM817190 |
UW.Fetal_Renal_Cortex_Left.ChromatinAccessibility.
H-24169.DS18931.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Cortex_Left.
H-24169 | Fetal Renal
Cortex | Hotspots | Fetal_Renal_
Cortex_Left | H-24169 | GSM817203 |
UW.Fetal_Renal_Cortex_Left.ChromatinAccessibility.
H-24232.DS19257.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Cortex_Left.
H-24232 | Fetal Renal
Cortex | Hotspots | Fetal_Renal_
Cortex_Left | H-24232 | GSM817211 |
UW.Fetal_Renal_Cortex_Right.ChromatinAccessibility.
H-23871.DS17545.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Cortex_Right.
H-23871 | Fetal Renal
Cortex | Hotspots | Fetal_Renal_
Cortex_Right | H-23871 | GSM701517 |
UW.Fetal_Renal_Cortex_Right.ChromatinAccessibility.
H-24169.DS18928.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Cortex_Right.
H-24169 | Fetal Renal
Cortex | Hotspots | Fetal_Renal_
Cortex_Right | H-24169 | GSM817202 |
UW.Fetal_Renal_Cortex_Right.ChromatinAccessibility.
H-24232.DS19254.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Cortex_Right.
H-24232 | Fetal Renal
Cortex | Hotspots | Fetal_Renal_
Cortex_Right | H-24232 | GSM817210 |
UW.Fetal_Renal_Pelvis.ChromatinAccessibility.H-23790.
DS17381.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Pelvis.H-23790 | Fetal Renal
Pelvis | Hotspots | Fetal_Renal_
Pelvis | H-23790 | GSM701501 |
UW.Fetal_Renal_Pelvis.ChromatinAccessibility.H-23833.
DS17451.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Pelvis.H-23833 | Fetal Renal
Pelvis | Hotspots | Fetal_Renal_
Pelvis | H-23833 | GSM701510 |
UW.Fetal_Renal_Pelvis.ChromatinAccessibility.H-23914.
DS17760.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Pelvis.H-23914 | Fetal Renal
Pelvis | Hotspots | Fetal_Renal_
Pelvis | H-23914 | GSM774222 |
UW.Fetal_Renal_Pelvis.ChromatinAccessibility.H-24005.
DS18088.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Pelvis.H-24005 | Fetal Renal
Pelvis | Hotspots | Fetal_Renal_
Pelvis | H-24005 | GSM774236 |
UW.Fetal_Renal_Pelvis.ChromatinAccessibility.H-24078.
DS18431.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Pelvis.H-24078 | Fetal Renal
Pelvis | Hotspots | Fetal_Renal_
Pelvis | H-24078 | GSM817177 |
UW.Fetal_Renal_Pelvis.ChromatinAccessibility.H-24259.
DS19386.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Pelvis.H-24259 | Fetal Renal
Pelvis | Hotspots | Fetal_Renal_
Pelvis | H-24259 | GSM817218 |
UW.Fetal_Renal_Pelvis.ChromatinAccessibility.H-24477.
DS20448.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Pelvis.H-24477 | Fetal Renal
Pelvis | Hotspots | Fetal_Renal_
Pelvis | H-24477 | GSM878662 |
UW.Fetal_Renal_Pelvis_Left.ChromatinAccessibility.
H-23871.DS17553.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Pelvis_Left.
H-23871 | Fetal Renal
Pelvis | Hotspots | Fetal_Renal_
Pelvis_Left | H-23871 | GSM701520 |
UW.Fetal_Renal_Pelvis_Left.ChromatinAccessibility.
H-24111.DS18666.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Pelvis_Left.
H-24111 | Fetal Renal
Pelvis | Hotspots | Fetal_Renal_
Pelvis_Left | H-24111 | GSM817193 |
UW.Fetal_Renal_Pelvis_Left.ChromatinAccessibility.
H-24169.DS18964.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Pelvis_Left.
H-24169 | Fetal Renal
Pelvis | Hotspots | Fetal_Renal_
Pelvis_Left | H-24169 | GSM817205 |
UW.Fetal_Renal_Pelvis_Left.ChromatinAccessibility.
H-24232.DS19238.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Pelvis_Left.
H-24232 | Fetal Renal
Pelvis | Hotspots | Fetal_Renal_
Pelvis_Left | H-24232 | GSM817209 |
UW.Fetal_Renal_Pelvis_Right.ChromatinAccessibility.
H-23871.DS17548.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Pelvis_Right.
H-23871 | Fetal Renal
Pelvis | Hotspots | Fetal_Renal_
Pelvis_Right | H-23871 | GSM701518 |
UW.Fetal_Renal_Pelvis_Right.ChromatinAccessibility.
H-24111.DS18663.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Pelvis_Right.
H-24111 | Fetal Renal
Pelvis | Hotspots | Fetal_Renal_
Pelvis_Right | H-24111 | GSM817192 |
UW.Fetal_Renal_Pelvis_Right.ChromatinAccessibility.
H-24169.DS18961.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Pelvis_Right.
H-24169 | Fetal Renal
Pelvis | Hotspots | Fetal_Renal_
Pelvis_Right | H-24169 | GSM817204 |
UW.Fetal_Renal_Pelvis_Right.ChromatinAccessibility.
H-24232.DS19235.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Renal_
Pelvis_Right.
H-24232 | Fetal Renal
Pelvis | Hotspots | Fetal_Renal_
Pelvis_Right | H-24232 | GSM817208 |
UW.Fetal_Skin.ChromatinAccessibility.H-22337.DS10987.
twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Skin.
H-22337 | Fetal Skin | Hotspots | Fetal_Skin | H-22337 | GSM817158 |
UW.Fetal_Spinal_Cord.ChromatinAccessibility.H-24111.
DS18501.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Spinal_
Cord.H-24111 | Fetal Spinal Cord | Hotspots | Fetal_Spinal_
Cord | H-24111 | GSM817189 |
UW.Fetal_Spinal_Cord.ChromatinAccessibility.H-24409.
DS20351.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Spinal_
Cord.H-24409 | Fetal Spinal Cord | Hotspots | Fetal_Spinal_
Cord | H-24409 | GSM878661 |
UW.Fetal_Spinal_Cord.ChromatinAccessibility.H-24493.
DS20530.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Spinal_
Cord.H-24493 | Fetal Spinal Cord | Hotspots | Fetal_Spinal_
Cord | H-24493 | GSM878663 |
UW.Fetal_Spleen.ChromatinAccessibility.H-23399.
DS17448.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Spleen.
H-23399 | Fetal Spleen | Hotspots | Fetal_Spleen | H-23399 | GSM701509 |
UW.Fetal_Stomach.ChromatinAccessibility.H-23589.
DS16530.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Stomach.
H-23589 | Fetal Stomach | Hotspots | Fetal_Stomach | H-23589 | GSM774202 |
UW.Fetal_Stomach.ChromatinAccessibility.H-23758.
DS17172.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Stomach.
H-23758 | Fetal Stomach | Hotspots | Fetal_Stomach | H-23758 | GSM774212 |
UW.Fetal_Stomach.ChromatinAccessibility.H-23769.
DS17325.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Stomach.
H-23769 | Fetal Stomach | Hotspots | Fetal_Stomach | H-23769 | GSM701498 |
UW.Fetal_Stomach.ChromatinAccessibility.H-23887.
DS17659.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Stomach.
H-23887 | Fetal Stomach | Hotspots | Fetal_Stomach | H-23887 | GSM701521 |
UW.Fetal_Stomach.ChromatinAccessibility.H-23914.
DS17750.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Stomach.
H-23914 | Fetal Stomach | Hotspots | Fetal_Stomach | H-23914 | GSM701528 |
UW.Fetal_Stomach.ChromatinAccessibility.H-23964.
DS17878.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Stomach.
H-23964 | Fetal Stomach | Hotspots | Fetal_Stomach | H-23964 | GSM701538 |
UW.Fetal_Stomach.ChromatinAccessibility.H-24005.
DS17963.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Stomach.
H-24005 | Fetal Stomach | Hotspots | Fetal_Stomach | H-24005 | GSM774232 |
UW.Fetal_Stomach.ChromatinAccessibility.H-24078.
DS18389.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Stomach.
H-24078 | Fetal Stomach | Hotspots | Fetal_Stomach | H-24078 | GSM817173 |
UW.Fetal_Stomach.ChromatinAccessibility.H-24125.
DS18821.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Stomach.
H-24125 | Fetal Stomach | Hotspots | Fetal_Stomach | H-24125 | GSM817199 |
UW.Fetal_Stomach.ChromatinAccessibility.H-24401.
DS20349.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Stomach.
H-24401 | Fetal Stomach | Hotspots | Fetal_Stomach | H-24401 | GSM878660 |
UW.Fetal_Stomach.ChromatinAccessibility.H-24507.
DS20546.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Stomach.
H-24507 | Fetal Stomach | Hotspots | Fetal_Stomach | H-24507 | GSM878665 |
UW.Fetal_Testes.ChromatinAccessibility.H24198_H23640_
H24042.DS18942.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Testes.
H24198_
H23640_H24042 | Fetal Testes | Hotspots | Fetal_Testes | H24198_
H23640_
H24042 | GSM878617 |
UW.Fetal_Thymus.ChromatinAccessibility.H-23589.
DS16490.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Thymus.
H-23589 | Fetal Thymus | Hotspots | Fetal_Thymus | H-23589 | GSM665823 |
UW.Fetal_Thymus.ChromatinAccessibility.H-23663.
DS16841.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Thymus.
H-23663 | Fetal Thymus | Hotspots | Fetal_Thymus | H-23663 | GSM774204 |
UW.Fetal_Thymus.ChromatinAccessibility.H-23769.
DS17323.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Thymus.
H-23769 | Fetal Thymus | Hotspots | Fetal_Thymus | H-23769 | GSM701497 |
UW.Fetal_Thymus.ChromatinAccessibility.H-23833.
DS17474.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Thymus.
H-23833 | Fetal Thymus | Hotspots | Fetal_Thymus | H-23833 | GSM701513 |
UW.Fetal_Thymus.ChromatinAccessibility.H-23964.
DS17875.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Thymus.
H-23964 | Fetal Thymus | Hotspots | Fetal_Thymus | H-23964 | GSM701537 |
UW.Fetal_Thymus.ChromatinAccessibility.H-23964.
DS17876.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Thymus.
H-23964 | Fetal Thymus | Hotspots | Fetal_Thymus | H-23964 | GSM774230 |
UW.Fetal_Thymus.ChromatinAccessibility.H-24078.
DS18382.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Thymus.
H-24078 | Fetal Thymus | Hotspots | Fetal_Thymus | H-24078 | GSM817172 |
UW.Fetal_Thymus.ChromatinAccessibility.H-24259.
DS19287.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Thymus.
H-24259 | Fetal Thymus | Hotspots | Fetal_Thymus | H-24259 | GSM878627 |
UW.Fetal_Thymus.ChromatinAccessibility.H-24401.
DS20335.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Thymus.
H-24401 | Fetal Thymus | Hotspots | Fetal_Thymus | H-24401 | GSM878656 |
UW.Fetal_Thymus.ChromatinAccessibility.H-24409.
DS20341.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fetal_Thymus.
H-24409 | Fetal Thymus | Hotspots | Fetal_Thymus | H-24409 | GSM878657 |
UW.Fibroblasts_Fetal_Skin_Abdomen.
ChromatinAccessibility.H-24259.DS19558.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fibroblasts_
Fetal_Skin_
Abdomen.
H-24259 | Fibroblast | Hotspots | Fibroblasts_
Fetal_Skin_
Abdomen | H-24259 | GSM878635 |
UW.Fibroblasts_Fetal_Skin_Abdomen.
ChromatinAccessibility.H-24259.DS19561.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fibroblasts_
Fetal_Skin_
Abdomen.
H-24259 | Fibroblast | Hotspots | Fibroblasts_
Fetal_Skin_
Abdomen | H-24259 | GSM878636 |
UW.Fibroblasts_Fetal_Skin_Back.ChromatinAccessibility.
H-24218.DS19233.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fibroblasts_Fetal_
Skin_Back.
H-24218 | Fibroblast | Hotspots | Fibroblasts_
Fetal_Skin_Back | H-24218 | GSM878624 |
UW.Fibroblasts_Fetal_Skin_Biceps_Left.
ChromatinAccessibility.H-24259.DS19857.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fibroblasts_
Fetal_Skin_
Biceps_Left.
H-24259 | Fibroblast | Hotspots | Fibroblasts_
Fetal_Skin_
Biceps_Left | H-24259 | GSM878644 |
UW.Fibroblasts_Fetal_Skin_Biceps_Left.
ChromatinAccessibility.H-24259.DS19867.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fibroblasts_
Fetal_Skin_
Biceps_Left.
H-24259 | Fibroblast | Hotspots | Fibroblasts_
Fetal_Skin_
Biceps_Left | H-24259 | GSM878645 |
UW.Fibroblasts_Fetal_Skin_Biceps_Right.
ChromatinAccessibility.H-24259.DS19745.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fibroblasts_
Fetal_Skin_
Biceps_Right.
H-24259 | Fibroblast | Hotspots | Fibroblasts_
Fetal_Skin_
Biceps_Right | H-24259 | GSM878642 |
UW.Fibroblasts_Fetal_Skin_Biceps_Right.
ChromatinAccessibility.H-24259.DS19761.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fibroblasts_
Fetal_Skin_
Biceps_Right.
H-24259 | Fibroblast | Hotspots | Fibroblasts_
Fetal_Skin_
Biceps_Right | H-24259 | GSM878643 |
UW.Fibroblasts_Fetal_Skin_Quadriceps_Left.
ChromatinAccessibility.H-24259.DS20046.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fibroblasts_
Fetal_Skin_
Quadriceps_
Left.H-24259 | Fibroblast | Hotspots | Fibroblasts_
Fetal_Skin_
Quadriceps_Left | H-24259 | GSM878648 |
UW.Fibroblasts_Fetal_Skin_Quadriceps_Left.
ChromatinAccessibility.H-24259.DS20056.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fibroblasts_
Fetal_Skin_
Quadriceps_
Left.H-24259 | Fibroblast | Hotspots | Fibroblasts_
Fetal_Skin_
Quadriceps_Left | H-24259 | GSM878649 |
UW.Fibroblasts_Fetal_Skin_Quadriceps_Right.
ChromatinAccessibility.H-24259.DS19943.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fibroblasts_
Fetal_Skin_
Quadriceps_
Right.H-24259 | Fibroblast | Hotspots | Fibroblasts_
Fetal_Skin_
Quadriceps_
Right | H-24259 | GSM878646 |
UW.Fibroblasts_Fetal_Skin_Quadriceps_Right.
ChromatinAccessibility.H-24259.DS19948.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fibroblasts_
Fetal_Skin_
Quadriceps_
Right.H-24259 | Fibroblast | Hotspots | Fibroblasts_
Fetal_Skin_
Quadriceps_
Right | H-24259 | GSM878647 |
UW.Fibroblasts_Fetal_Skin_Scalp.ChromatinAccessibility.
H-24259.DS19444.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fibroblasts_
Fetal_Skin_
Scalp.H-24259 | Fibroblast | Hotspots | Fibroblasts_
Fetal_Skin_Scalp | H-24259 | GSM878633 |
UW.Fibroblasts_Fetal_Skin_Scalp.ChromatinAccessibility.
H-24259.DS19449.twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fibroblasts_
Fetal_Skin_
Scalp.H-24259 | Fibroblast | Hotspots | Fibroblasts_
Fetal_Skin_Scalp | H-24259 | GSM878634 |
UW.Fibroblasts_Fetal_Skin_Upper_Back.
ChromatinAccessibility.H-24259.DS19696.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fibroblasts_
Fetal_Skin_
Upper_Back.
H-24259 | Fibroblast | Hotspots | Fibroblasts_
Fetal_Skin_
Upper_Back | H-24259 | GSM878640 |
UW.Fibroblasts_Fetal_Skin_Upper_Back.
ChromatinAccessibility.H-24259.DS19706.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Fibroblasts_
Fetal_Skin_
Upper_Back.
H-24259 | Fibroblast | Hotspots | Fibroblasts_
Fetal_Skin_
Upper_Back | H-24259 | GSM878641 |
UW.H1_BMP4_Derived_Mesendoderm_Cultured_Cells.
ChromatinAccessibility.DS18732.twopass.merge150.
wgt10.zgt2.wig | UW | DNase-seq | Roadmap | H1_BMP4_
Derived_
Mesendoderm_
Cultured_Cells | ES Cell | Hotspots | Mesendoderm_
Cultured_Cells | NA | GSM817198 |
UW.H1_BMP4_Derived_Mesendoderm_Cultured_Cells.
ChromatinAccessibility.DS19310.twopass.merge150.
wgt10.zgt2.wig | UW | DNase-seq | Roadmap | H1_BMP4_
Derived_
Mesendoderm_
Cultured_Cells | ES Cell | Hotspots | Mesendoderm_
Cultured_Cells | NA | GSM817215 |
UW.H1_BMP4_Derived_Trophoblast_Cultured_Cells.
ChromatinAccessibility.DS18736.twopass.merge150.
wgt10.zgt2.wig | UW | DNase-seq | Roadmap | H1_BMP4_
Derived_
Trophoblast_
Cultured_Cells | ES Cell | Hotspots | Trophoblast_
Cultured_Cells | NA | GSM878614 |
UW.H1_BMP4_Derived_Trophoblast_Cultured_Cells.
ChromatinAccessibility.DS19317.twopass.merge150.
wgt10.zgt2.wig | UW | DNase-seq | Roadmap | H1_BMP4_
Derived_
Trophoblast_
Cultured_Cells | ES Cell | Hotspots | Trophoblast_
Cultured_Cells | NA | GSM878628 |
UW.H1.ChromatinAccessibility.DS18873.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | H1 | ES Cell | Hotspots | H1 | NA | GSM878616 |
UW.H1.ChromatinAccessibility.DS19100.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | H1 | ES Cell | Hotspots | H1 | NA | GSM878621 |
UW.H1_Derived_Mesenchymal_Stem_Cells.
ChromatinAccessibility.DS20671.twopass.merge150.
wgt10.zgt2.wig | UW | DNase-seq | Roadmap | H1_Derived_
Mesenchymal_
Stem_Cells | ES Cell | Hotspots | Mesenchymal_
Stem_Cells | NA | GSM906380 |
UW.H1_Derived_Mesenchymal_Stem_Cells.
ChromatinAccessibility.DS21042.twopass.merge150.
wgt10.zgt2.wig | UW | DNase-seq | Roadmap | H1_Derived_
Mesenchymal_
Stem_Cells | ES Cell | Hotspots | Mesenchymal_
Stem_Cells | NA | GSM906381 |
UW.H1_Derived_Neuronal_Progenitor_Cultured_Cells.
ChromatinAccessibility.DS18739.twopass.merge150.
wgt10.zgt2.wig | UW | DNase-seq | Roadmap | H1_Derived_
Neuronal_
Progenitor_
Cultured_Cells | ES Cell | Hotspots | Neuronal_
Progenitor_
Cultured_Cells | NA | GSM878615 |
UW.H1_Derived_Neuronal_Progenitor_Cultured_Cells.
ChromatinAccessibility.DS20153.twopass.merge150.
wgt10.zgt2.wig | UW | DNase-seq | Roadmap | H1_Derived_
Neuronal_
Progenitor_
Cultured_Cells | ES Cell | Hotspots | Neuronal_
Progenitor_
Cultured_Cells | NA | GSM906379 |
UW.H9.ChromatinAccessibility.DS18517.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | H9 | ES cell | Hotspots | H9 | NA | GSM878612 |
UW.H9.ChromatinAccessibility.DS18522.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | H9 | ES cell | Hotspots | H9 | NA | GSM878613 |
UW.IMR90.ChromatinAccessibility.DS11759.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | IMR90 | Lung | Hotspots | IMR90 | NA | GSM468792 |
UW.IMR90.ChromatinAccessibility.DS11764.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | IMR90 | Lung | Hotspots | IMR90 | NA | GSM468801 |
UW.IMR90.ChromatinAccessibility.DS13219.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | IMR90 | Lung | Hotspots | IMR90 | NA | GSM530665 |
UW.IMR90.ChromatinAccessibility.DS13229.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | IMR90 | Lung | Hotspots | IMR90 | NA | GSM530666 |
UW.iPS_DF_19.11.ChromatinAccessibility.DS15153.
twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | iPS_DF_19.11 | IPS cell | Hotspots | iPS_DF_19.11 | NA | GSM665801 |
UW.iPS_DF_19.7.ChromatinAccessibility.DS15148.
twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | iPS_DF_19.7 | IPS cell | Hotspots | iPS_DF_19.7 | NA | GSM665800 |
UW.iPS_DF_4.7.ChromatinAccessibility.DS15169.
twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | iPS_DF_4.7 | IPS cell | Hotspots | iPS_DF_4.7 | NA | GSM665803 |
UW.iPS_DF_6.9.ChromatinAccessibility.DS15164.
twopass.merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | iPS_DF_6.9 | IPS cell | Hotspots | iPS_DF_6.9 | NA | GSM665802 |
UW.Mobilized_CD34_Primary_Cells.
ChromatinAccessibility.RO_00738.DS13199.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Mobilized_CD34_
Primary_Cells.
RO_00738+ | Blood | Hotspots | CD34_Mobilised; | RO_00738+ | GSM530664 |
UW.Mobilized_CD34_Primary_Cells.
ChromatinAccessibility.RO_01480.DS12771.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Mobilized_CD34_
Primary_Cells.
RO_01480+ | Blood | Hotspots | CD34_Mobilised; | RO_01480+ | GSM530658 |
UW.Mobilized_CD34_Primary_Cells.
ChromatinAccessibility.RO_01492.DS12774.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Mobilized_CD34_
Primary_Cells.
RO_01492+ | Blood | Hotspots | CD34_Mobilised; | RO_01492+ | GSM530659 |
UW.Mobilized_CD34_Primary_Cells.
ChromatinAccessibility.RO_01492.DS14206.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Mobilized_CD34_
Primary_Cells.
RO_01492+ | Blood | Hotspots | CD34_Mobilised; | RO_01492+ | GSM595919 |
UW.Mobilized_CD34_Primary_Cells.
ChromatinAccessibility.RO_01508.DS11202.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Mobilized_CD34_
Primary_Cells.
RO_01508+ | Blood | Hotspots | CD34_Mobilised; | RO_01508+ | GSM493386 |
UW.Mobilized_CD34_Primary_Cells.
ChromatinAccessibility.RO_01517.DS14129.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Mobilized_CD34_
Primary_Cells.
RO_01517+ | Blood | Hotspots | CD34_Mobilised; | RO_01517+ | GSM595917 |
UW.Mobilized_CD34_Primary_Cells.
ChromatinAccessibility.RO_01520.DS12785.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Mobilized_CD34_
Primary_Cells.
RO_01520+ | Blood | Hotspots | CD34_Mobilised; | RO_01520+ | GSM530660 |
UW.Mobilized_CD34_Primary_Cells.
ChromatinAccessibility.RO_01527.DS11666.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Mobilized_CD34_
Primary_Cells.
RO_01527+ | Blood | Hotspots | CD34_Mobilised; | RO_01527+ | GSM493387 |
UW.Mobilized_CD34_Primary_Cells.
ChromatinAccessibility.RO_01535.DS12274.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Mobilized_CD34_
Primary_Cells.
RO_01535+ | Blood | Hotspots | CD34_Mobilised; | RO_01535+ | GSM493384 |
UW.Mobilized_CD34_Primary_Cells.
ChromatinAccessibility.RO_01535.DS14197.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Mobilized_CD34_
Primary_Cells.
RO_01535+ | Blood | Hotspots | CD34_Mobilised; | RO_01535+ | GSM595918 |
UW.Mobilized_CD34_Primary_Cells.
ChromatinAccessibility.RO_01536.DS12339.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Mobilized_CD34_
Primary_Cells.
RO_01536+ | Blood | Hotspots | CD34_Mobilised; | RO_01536+ | GSM530652 |
UW.Mobilized_CD34_Primary_Cells.
ChromatinAccessibility.RO_01549.DS12734.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Mobilized_CD34_
Primary_Cells.
RO_01549+ | Blood | Hotspots | CD34_Mobilised; | RO_01549+ | GSM530657 |
UW.Mobilized_CD34_Primary_Cells.
ChromatinAccessibility.RO_01562.DS13196.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Mobilized_CD34_
Primary_Cells.
RO_01562+ | Blood | Hotspots | CD34_Mobilised; | RO_01562+ | GSM530663 |
UW.Mobilized_CD34_Primary_Cells.
ChromatinAccessibility.RO_01701.DS17112.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Mobilized_CD34_
Primary_Cells.
RO_01701+ | Blood | Hotspots | CD34_Mobilised; | RO_01701+ | GSM774208 |
UW.Mobilized_CD4_Primary_Cells.
ChromatinAccessibility.RO_01679.DS16955.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Mobilized_CD4_
Primary_Cells.
RO_01679+ | Blood | Hotspots | CD4_Mobilised; | RO_01679+ | GSM701489 |
UW.Mobilized_CD4_Primary_Cells.
ChromatinAccessibility.RO_01701.DS17175.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Mobilized_CD4_
Primary_Cells.
RO_01701+ | Blood | Hotspots | CD4_Mobilised; | RO_01701+ | GSM701491 |
UW.Mobilized_CD56_Primary_Cells.
ChromatinAccessibility.RO_01689.DS16376.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Mobilized_CD56_
Primary_Cells.
RO_01689+ | Blood | Hotspots | CD56_Mobilised; | RO_01689+ | GSM665820 |
UW.Mobilized_CD8_Primary_Cells.
ChromatinAccessibility.RO_01679.DS16962.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Mobilized_CD8_
Primary_Cells.
RO_01679+ | Blood | Hotspots | CD8_Mobilised; | RO_01679+ | GSM817160 |
UW.Penis_Foreskin_Fibroblast_Primary_Cells.
ChromatinAccessibility.skin01.DS18224.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Penis_Foreskin_
Fibroblast_
Primary_Cells.
skin01 | Fibroblast | Hotspots | Penis_Foreskin_
Fibroblast | skin01 | GSM774240 |
UW.Penis_Foreskin_Fibroblast_Primary_Cells.
ChromatinAccessibility.skin01.DS18229.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Penis_Foreskin_
Fibroblast_
Primary_Cells.
skin01 | Fibroblast | Hotspots | Penis_Foreskin_
Fibroblast | skin01 | GSM774241 |
UW.Penis_Foreskin_Fibroblast_Primary_Cells.
ChromatinAccessibility.skin02.DS18252.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Penis_Foreskin_
Fibroblast_
Primary_Cells.
skin02 | Fibroblast | Hotspots | Penis_Foreskin_
Fibroblast | skin02 | GSM817169 |
UW.Penis_Foreskin_Fibroblast_Primary_Cells.
ChromatinAccessibility.skin02.DS18256.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Penis_Foreskin_
Fibroblast_
Primary_Cells.
skin02 | Fibroblast | Hotspots | Penis_Foreskin_
Fibroblast | skin02 | GSM817170 |
UW.Penis_Foreskin_Keratinocyte_Primary_Cells.
ChromatinAccessibility.skin01.DS18692.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Penis_Foreskin_
Keratinocyte_
Primary_Cells.
skin01 | Skin | Hotspots | Penis_Foreskin_
Keratinocyte | skin01 | GSM817194 |
UW.Penis_Foreskin_Keratinocyte_Primary_Cells.
ChromatinAccessibility.skin01.DS18695.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Penis_Foreskin_
Keratinocyte_
Primary_Cells.
skin01 | Skin | Hotspots | Penis_Foreskin_
Keratinocyte | skin01 | GSM817195 |
UW.Penis_Foreskin_Keratinocyte_Primary_Cells.
ChromatinAccessibility.skin02.DS18714.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Penis_Foreskin_
Keratinocyte_
Primary_Cells.
skin02 | Skin | Hotspots | Penis_Foreskin_
Keratinocyte | skin02 | GSM817196 |
UW.Penis_Foreskin_Keratinocyte_Primary_Cells.
ChromatinAccessibility.skin02.DS18718.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Penis_Foreskin_
Keratinocyte_
Primary_Cells.
skin02 | Skin | Hotspots | Penis_Foreskin_
Keratinocyte | skin02 | GSM817197 |
UW.Penis_Foreskin_Melanocyte_Primary_Cells.
ChromatinAccessibility.skin01.DS18590.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Penis_Foreskin_
Melanocyte_
Primary_Cells.
skin01 | Skin | Hotspots | Penis_Foreskin_
Melanocyte | skin01 | GSM774243 |
UW.Penis_Foreskin_Melanocyte_Primary_Cells.
ChromatinAccessibility.skin01.DS18601.twopass.
merge150.wgt10.zgt2.wig | UW | DNase-seq | Roadmap | Penis_Foreskin_
Melanocyte_
Primary_Cells.
skin01 | Skin | Hotspots | Penis_Foreskin_
Melanocyte | skin01 | GSM774244 |
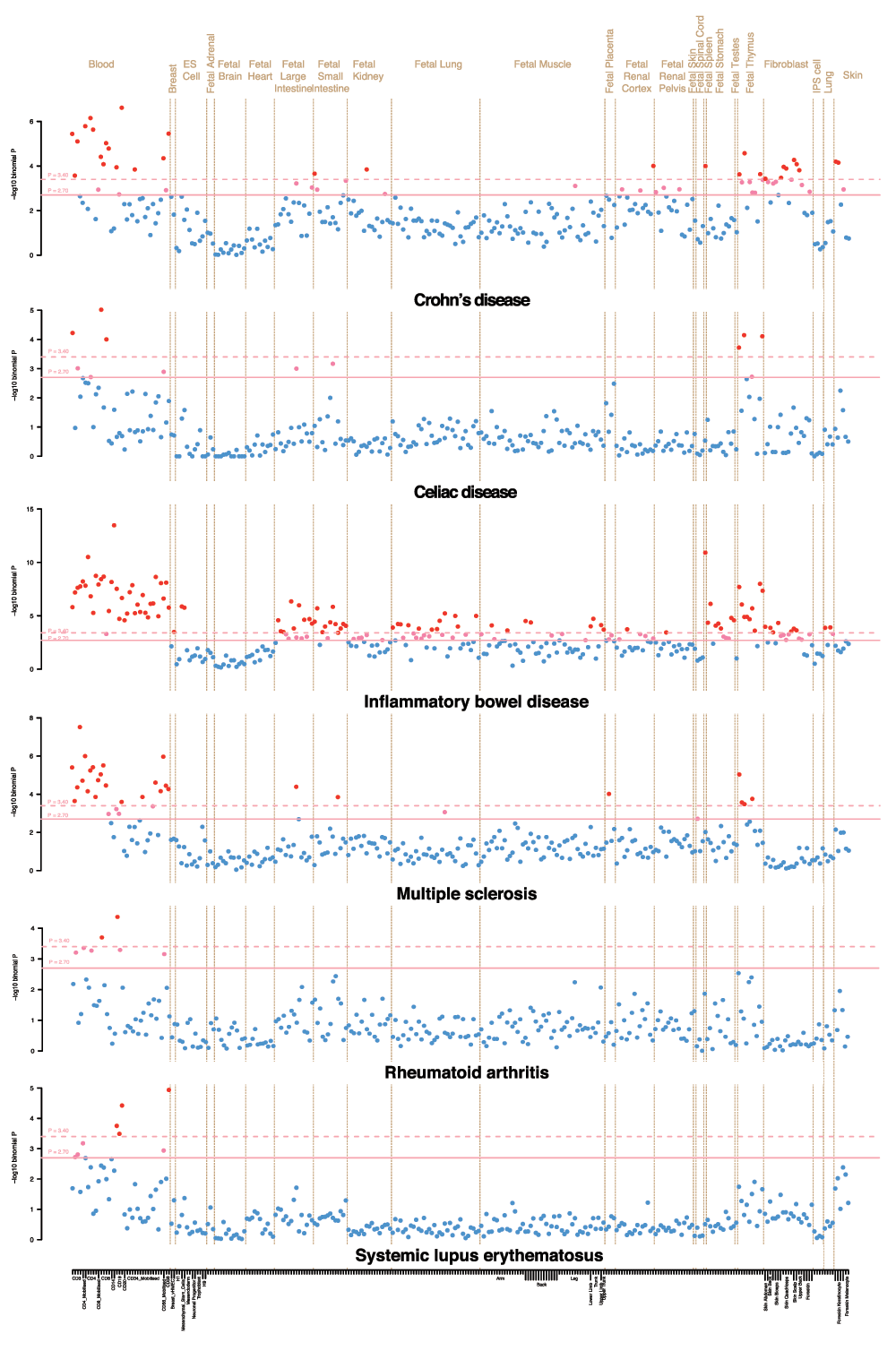
There is also a related ambiguity to the labelling of the threshold options in the web tool, which I will get fixed shortly.
There is also a related ambiguity to the labelling of the threshold options in the web tool, which I will get fixed shortly.