Keywords
microbiome, metagenomics, neurological infections, computational biology, next-generation sequencing, sequence alignment
This article is included in the Preclinical Reproducibility and Robustness gateway.
microbiome, metagenomics, neurological infections, computational biology, next-generation sequencing, sequence alignment
We have revised our manuscript to make some small text amendments, in response to Charles Chiu's comments.
To read any peer review reports and author responses for this article, follow the "read" links in the Open Peer Review table.
Metagenomic shotgun sequencing, in which DNA or RNA is extracted from a tissue sample and then sequenced, has the potential to detect a wide range of infections. Deep whole-genome shotgun (WGS) sequencing can detect bacteria, viruses, and eukaryotic pathogens with equal effectiveness, as long as the infectious agent is similar to a species that has been previously sequenced. Sequencing databases already contain thousands of known species, and as this number grows, the sensitivity of WGS will grow as well.
In 2014, a large outbreak of infection with enterovirus D68 was associated with both severe respiratory illness and acute paralysis, which the U.S. Centers for Disease Control and Prevention (CDC) named acute flaccid myelitis (AFM)1. Samples collected from 48 patients were sequenced and shown to form a novel strain, Clade B1, based on phylogenetic analysis of 180 complete enterovirus D68 sequences2. The same study conducted metagenomic sequencing of cerebrospinal fluid (CSF) and/or nasopharyngeal (NP) swabs from 22 of these patients and found enterovirus D68 in some NP samples that were positive based on PCR testing.
The identification of species from a WGS sample is a challenging problem that has spurred the development of multiple new computational methods3–5. Because of the large size of next-generation sequencing data sets, these methods need to be very fast, but in the context of clinical diagnosis, they also need to be accurate. We downloaded the 31 next-generation sequencing (NGS) samples from the Greninger et al.2 study (NCBI accession SRP055445) and re-analyzed them using a computational pipeline based on the recently developed Kraken metagenomic analysis software4, a very fast and sensitive system that can be customized to use a database containing any species whose sequences are available.
Among the 22 subjects for which NGS data were available, we found at least two that had far greater numbers of sequences (reads) from a bacterial pathogen than from enterovirus D68. Neither subject had been reported in 2 as having a bacterial infection.
In one subject, US/CA/09-871, reported by Greninger et al.2 as positive for enterovirus D68 through PCR and metagenomic NGS, we found in the NP swab sample an overwhelming presence of bacterial sequences from Haemophilus influenzae, a known cause of meningitis and neurological complications that was a common infection prior to the development of an effective vaccine.
Specifically, we identified 2,389,621 reads from H. influenzae in this subject, with the closest similarity to strain R2846. These reads comprise 93% of all microbial reads identified at the species level in the sample. Greninger et al.2 reported 2,742 reads (in their Supplementary Table 4) matching enterovirus D682 but did not report finding any H. influenzae reads from this sample. Our analysis found 1,330 reads matching enterovirus D68.
To confirm the identity of these reads, we aligned them separately to the complete genome of H. influenzae R2846, and we found that the reads completely covered the genome. Dividing the genome into 100 kilobase windows, depth of coverage varied from 266–828 reads/100Kbp, with far deeper coverage as expected at the 16S ribosomal RNA genes.
The enterovirus D68 isolated from patient US/CA/09-871 differed from the others in that it appeared in 2009, well before the 2014 outbreak, and that it grouped with Clade C, phylogenetically distinct from Clade B1 that was associated with AFM. This patient was reported2 as having respiratory illness but not AFM. The sequence evidence here suggests that the patient might have had complications from H. influenzae-associated infection, although no clinical or CSF data was available for our re-analysis.
In a second subject, US/CA/12-5837, we found a strikingly large number of reads from Staphylococcus aureus in the NP swabs. The two separate NGS files associated with this subject contained 6,858,453 and 1,343,806 reads, comprising 70% and 84% (respectively) of all non-human reads identified at the species level in each sample. The closest match was S. aureus subsp. aureus MRSA252, a methicillin-resistant strain. The coverage was deep enough, approximately 40X, that it would be possible to assemble this genome separately from the reads here (Figure 1). Greninger et al.2 reported 2,790 reads from enterovirus D68 in this subject (our analysis found 1,641) but did not report any from S. aureus.
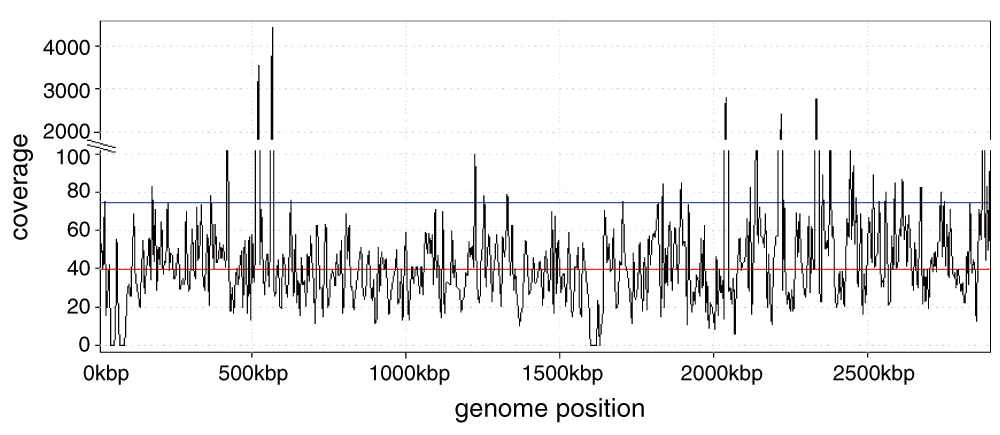
High peaks correspond to 16S rRNA genes. Red line: median coverage; blue line: mean coverage.
Patient US/CA/12-5837 was sampled in 2012, two years before the outbreak of AFM, although this patient was described in Greninger et al.2 as positive for enterovirus D68 based on clinical PCR testing and metagenomic sequencing. This patient is reported to be one of the first patients with enterovirus-D68-positive AFM2, but the sequence evidence indicates a severe S. aureus infection that might explain at least some of the patient’s symptoms. S. aureus has been implicated in neurological complications such as myelitis6 and meningitis7 by mechanisms that involve not only direct invasion into the central nervous system (CNS), but also immunopathogenic responses triggered by superantigens that can target the CNS8. At a minimum, S. aureus infection was overlooked by the previous analysis. Although the potential role of bacterial infection in the neurological disease that affected these two subjects is difficult to assess because of the lack of clinical and CSF information, its involvement as a pathogenic co-factor should be evaluated.
The metagenomics data (NCBI accession SRP055445) released by Greninger et al.2 comprise 43 files which cover 22 of the 48 subjects from their study (in their Supplementary Table 1); the study did not conduct NGS for all subjects. Our metagenomics pipeline identifies human reads at the same time that it searches for pathogens; therefore we scanned the data for human as well as microbial content. Greninger et al.2 reported that all human sequences had been removed from these files. We found, however, that all samples contained large numbers of human reads, ranging from a low of 18,215 to a high of 6,159,868. These comprised as few as 0.5% to as many as 95.6% of the reads in each sample, as shown in Table 1.
Shown are the number of reads in each sample that clearly match the human genome and do not match any microbial species. AFM: acute flaccid myelits; NP: nasopharyngeal swap; CSF: cerebrospinal fluid.
The inclusion of human sequence data in the files deposited at NCBI was likely a result of a computational method (SURPI5) that was insufficiently sensitive. Although the exact cause cannot be determined here, it is well known that sequence alignment algorithms often trade speed for sensitivity; e.g., by allowing fewer mismatches, an aligner can process reads at a much higher rate, at the cost of missing some alignments. It is less clear why the very large numbers of matches to two bacteria were missed; for both these bacteria, complete genomes from multiple strains are available in GenBank. We used both the Kraken system4 and the Bowtie2 aligner9 to ensure both sensitivity and speed in our analysis.
Release of sequence data is highly valuable, if not essential, for reproducibility and validation of sequencing-based studies. Failure to filter human reads from a sample is not uncommon; a recent study10 found that Human Microbiome Project samples, from which human DNA was supposed to have been removed, contain up to 95% human sequence. This suggests that future efforts to deposit microbiome data need to employ more sensitive computational screens in order to avoid the unintentional release of human sequence data.
Sequences were extracted from SRP055445 and each file was separately run through the Kraken program version 0.10.6-beta (https://github.com/DerrickWood/kraken)4, which identifies species by comparison with a database of all 31-bp sequences in all species. The database included the human genome (version GRCh38.p2), all complete bacterial and viral genomes, selected fungal pathogens, and known laboratory vector sequences from the NCBI UniVec database (http://www.ncbi.nlm.nih.gov/tools/vecscreen/univec). Percentages of bacterial and viral reads in each sample were re-computed after excluding human and vector sequences. Reads matching more than one species were classified at the genus level or above. Reads from H. influenzae and S. aureus were re-aligned using Bowtie2 version 2.2.59, a very fast and sensitive program for alignment of NGS reads to a reference genome, with the --local option. Bowtie2 was also used to re-align all reads from US/CA/12-5837 and US/CA/09-871 to the sequence of multiple enterovirus D68 strains (GenBank accessions JX101846.1, AY426531.1, KM851231.1, KM892500.1, KM892501.1, KM881710.2, KP745751.1, KP745755.1, KP745757.1, KP745760.1, KP745764.1, KP745766.1, and KP745767.1). We report the highest number of reads matching any one of these strains.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Table S3: summary table of NGS read counts (bacterial, viral, other from both CSF and NP)
Table S4: ... Continue reading We would like to clarify the tables in the Supplementary Appendix of our paper:
Table S3: summary table of NGS read counts (bacterial, viral, other from both CSF and NP)
Table S4: summary table of viral read counts (CSF and NP)
Table S5: summary table of bacterial read counts (CSF only; our rationale for not including the bacterial data has been previously given)
Table S6: summary table of fungal and parasitic read counts (CSF only)
We would be happy to provide the tables corresponding to bacterial, fungal, and parasitic read counts in the NP swabs that were generated from our analysis upon request. In hindsight, it may have been better to include these tables in the original publication as part of the supplementary files.
Table S3: summary table of NGS read counts (bacterial, viral, other from both CSF and NP)
Table S4: summary table of viral read counts (CSF and NP)
Table S5: summary table of bacterial read counts (CSF only; our rationale for not including the bacterial data has been previously given)
Table S6: summary table of fungal and parasitic read counts (CSF only)
We would be happy to provide the tables corresponding to bacterial, fungal, and parasitic read counts in the NP swabs that were generated from our analysis upon request. In hindsight, it may have been better to include these tables in the original publication as part of the supplementary files.