Keywords
Next Generation sequencing, genome assembly, Cloud Computing
Next Generation sequencing, genome assembly, Cloud Computing
The earliest landmark in genome sequencing is that of the Bacteriophage MS21 between 1972 and 1976. Following that, two DNA sequencing techniques for longer DNA molecules were invented, first the Maxam-Gilbert2 (chemical cleavage) method and then the Sanger3 (or dideoxy) method. The Maxam-Gilbert method was based on nucleotide-specific cleavage by chemicals, and is best applied to oligonucleotides that and short sequences usually smaller than 50 base-pairs in length. On the other hand, the Sanger method was more widely used because it leveraged the Polymerase Chain Reaction (PCR) for automation of the technique, which also allows to sequence long strands of DNA including entire genes. In more detail, the Sanger technique is based on chain termination by dideoxynucleotides triphosphate (ddNTPs) during PCR elongation reactions (review in4). Although the automated Sanger method had dominated the industry for almost two decades, with sequencing applications and broad demand for the technology in genome variation studies, comparative genomics, evolution, forensics, diagnostic and applied therapeutics, it was still limiting due to its high cost and labor intensive process5.
The high demand for low-cost sequencing led to the development of high-throughput technologies also known as Next Generation Sequencing (NGS), that parallelize the sequencing process and lower the cost per sequenced DNA base-pair. NGS techniques achieve this by automating template preparation and using high-speed, precision fluorescence imaging, for highly parallel identification of the nucleotides. NGS technologies involve sequencing of a dense array of DNA fragments by iterative cycles of enzymatic manipulation and imaging-based data collection. The major NGS platforms are Roche/454FLX (http://454.com/), Illumina/Solexa Genome Analyzer (http://www.illumina.com/systems/genome_analyzer_iix.ilmn), Applied Biosystems SOLiD system (http://www.appliedbiosystems.com/absite/us/en/home/applications-technologies/solid-next-generation-sequencing.html), Helicos Heliscope (http://www.helicosbio.com/Products/HelicosregGeneticAnalysisSystem/HeliScopetradeSequencer/tabid/87/Default.aspx) and Pacific Biosciences (http://www.pacificbiosciences.com/). Summary statistics for the throughput and characteristics of these NGS platforms are shown Figure 1, while additional review studies are available in the literature6–8.

Although these platforms are quite diverse in sequencing biochemistry as well as in how the array is generated, their workflows are conceptually similar. First, a library is prepared by fragmenting PCR-amplified DNA randomly, followed by in vitro ligation to a common adaptor, that is small DNA oligonucleotide with known sequence. In the next step, clustered amplicons that are multiple copies of a single fragment are generated and serve as the sequencing fragments. This can be achieved by several approaches, including in situ polonies9, emulsion10 or bridge PCR11,12. Common to these methods is that PCR amplicons derived from any given single library molecule end up spatially clustered, either to a single location on a planar substrate (in situ polonies, bridge PCR), or to the surface of micron-scale beads, which can be recovered and arrayed (emulsion PCR).
The advantage of the NGS platforms is sequencing using single, amplified DNA fragments, avoiding the need for cloning required by the Sanger method. This also makes the technology applicable to genomes of un-cultivated microorganism populations, such as for example in metagenomics. A disadvantage of the new technology is that sequence data is in the form of short reads, presenting a challenge to developers of software and genome assembly algorithms. More specifically, it can be difficult to correctly assign reads and separate between genomic regions that contain sequence repeats even if using high-stringency alignments, especially if the lengths of the repeats are longer than the reads. In addition, repeat resolution and read alignments are further complicated by sequencing error, and therefore genome assembly software must tolerate imperfect sequence alignments. A reduced alignment stringency can return many false positives that results in chimeric assemblies where distant regions of the genome are mistakenly assembled together. The limitation of short reads is compensated by multiple overlaps of the sequenced DNA fragments, resulting in many times coverage of the genome sequence. Finally, genome assembly is hindered by regions that have nucleotide composition for which PCR does not achieve its optimum yield, resulting in non-uniform genome coverage that in turn leads to gaps in the assembly.
To alleviate some of these problems in genome assemblies with short reads, “paired-end” reads are used that are generated by a simple modification to the standard sequencing template preparation, in order to get the forward and reverse strands at the two ends of a DNA fragment. The unique paired-end sequencing protocol allows the user to choose the length of the insert (200–500 bp) and use the positional and distance information of the reads for validating the genome assembly, allowing for resolution of alignment ambiguities and chimeric assemblies.
Two approaches widely used in NGS genome assemblers are the Overlap/Layout/Consensus (OLC)13 and the de Bruijn Graph (DBG)14 method, both using K-mers as the basis for read alignment. A graph is an abstraction used in computer science, and is composed of “nodes” connected by “edges”. If the edges connecting the nodes can be traversed in one direction, the graph is a directed graph, whereas if the edges can be traversed in both the directions the graph is bi-directed15.
The OLC approach for genome assembly is the typical method for Sanger datasets, and is implemented in software such as Celera Assembler, PCAP and ARACHNE16–18. With this approach reads are represented as nodes in the graph, and nodes for overlapping reads are connected by an edge. In more detail, OLC assembles proceed in three phases: in the first phase overlap discovery involves all-against-all comparison, pair-wise read alignment. The algorithm used for that purpose is a seed and extend heuristic algorithm that pre-computes K-mers from all reads, then selects overlap candidate reads that share K-mers and calculates alignments using the K-mers as alignment seeds. This step of the algorithm is sensitive to settings of K-mer size, minimum overlap length and percent identity required to retain an overlap as true positive, in addition to base calling errors and low-coverage sequencing. Using the results from the read alignment, an overlap graph is constructed that provides an approximate read layout. The overlap graph does not include the sequence base calls rather than just the overlapping read identifiers, so large-genome graphs may fit into practical amounts of computer memory. The graph also has metadata on the nodes and edges, in order to distinguish the lengths, 5′ and 3′ ends, forward and reverse complements, and the type of overlap between the reads. In the second phase, the precise layout and the consensus sequence of the graph are determined by performing Multiple Sequence Alignment (MSA)19. For that purpose, the algorithm must load all the sequences into memory, and this becomes one of the most computationally demanding stages of the assembly. Finally, in the third phase the assembly algorithm follows a Hamiltonian path20 to “walk” through the graph visiting every node only once, and the contigs formed by merging the overlapping reads are determined.
The second approach in genome assembly is based on the de Brujn Graph (DBG) algorithms, and example software using this approach includes Velvet, Contrail, ALLPATHS and SOAPdenovo21–24. In the DBG approach each node in the graph corresponds to a unique K-mer present in the set of reads, and a directed edge connects two nodes labeled ‘a’ and ‘b’, if the k – 1 length suffix of ‘a’ is the same as the k – 1 length prefix of ‘b’. For example, a graph node representing the K-mer (K=3) ATG will have an edge with the node representing TGG. The DBG algorithms are more efficient for large datasets, as they doe not require all-against-all read alignment and overlap discovery, while it also doe not store individual reads or their overlaps in the computer memory. A de Brujn graph can either be uni-directed or bi- directed, with a single edge connecting two nodes, or edges with two directions for the 5’ or 3’ genome strands. A bidirected graph has the advantage of allowing simultaneous assembly of sequence reads from both strands of the genome15. The DBG assembly algorithms traverse the graph using an Eularian path14, where each edge is visited exactly once.
Assembly dataset: The sequence reads dataset were Illumina reads from the fish species M. zebrafish downloaded from Assemblathon database (http://assemblathon.org). The genome was approximately 1GB in size, the total library coverage estimate for this dataset was 197X. The read length for the given dataset was 101 and in the first phase 38,364,464 reads with coverage of only 2X, both paired and unpaired, totaling 2,762,241,408 base pairs were used (filtered for bad quality reads). Following that, different variations in the size of dataset used for running the assembly involving 1/4, 1/2 and the entire dataset of fish genome. The dataset consisted of pairs of files with each corresponding to the paired-end reads. De novo assembly was performed using two different assemblers, Velvet and Contrail, both using the graph-theoretic framework of De-Brujn Graph. Velvet requires high performance computer servers with large RAM memory, whereas Contrail is a distributed memory assembler that performs the computation in parallel over several servers on a computer cluster.
Assembly statistics: The output of the assembly was a file with a list of contigs of various lengths. The file contains contigs along with various details like length and coverage. For the Velvet assembler, the output file was parsed using a Perl script called velvet_stats.pl to calculate various assembly statistics. Similarly, for the Contrail assembler a Perl script called contrail_stats.pl was used. These scripts are available upon request from the authors. Using the Perl scripts the following statistics were calculated for comparing the quality of the two assemblies:
1) N50 score: The length of the largest contig for which the following is true: the sum of its length and the lengths of all larger contigs equals to 50% of the total contig length. The N50 size is computed by sorting all contigs from largest to smallest and by determining the minimum set of contigs whose sizes total 50% of the entire genome.
2) Maximum contig length: Contig with largest number basepairs.
3) Minimum contig length: Contig with smallest number of basepairs.
4) Mean contig length: Average of all the contigs length.
To the run the assembly on the full dataset using contrail, all the read files were merged into a single file, and copied in a Hadoop compute cluster available at JCVI that consisted of 8 servers with 32 GB RAM and 16cores each.
Both the Velvet and Contrail assemblers are designed for short reads. Velvet requires a compute server with large RAM memory, whereas Contrail relies on Hadoop to distribute the assembly graphs across multiple servers. Velvet is a de novo genomic assembler specially designed for short read sequencing technologies, such as Solexa or 454. It currently takes in short read sequences, removes errors then produces high quality, unique contigs using paired-end read and long read information when available, for resolving genomic repeats that complicate contig calculation. Velvet resolves errors which can arise due to both the sequencing process or to the polymorphisms by removing the “tips” in the de Bruijn graph that are chain of nodes that is disconnected on one end, or “bubbles” using the Tour Bus algorithm, both originating from nucleotide differences due to sequencing error or polymorphisms in diploid genomes.
Contrail enables de novo assembly of large genomes from short reads by leveraging Hadoop, a software library and framework that allows distributed processing of large data sets across clusters of computers using a simple programming model. The Hadoop Distributed File System (HDFS) is the primary storage system in this framework, and creates multiple data blocks distributed across compute server throughout a cluster to enable reliable, extremely rapid parallel computations. HDFS is highly fault-tolerant and is designed to be deployed on low-cost, commodity hardware. Build on top of HDFS is the Hadoop-MapReduce Framework that uses a “Map” step in which individual data records tagged with a “key” are processed in parallel. In a second step, a “Reduce” function performs aggregation and summarization, in which all associated records based on the key are brought together from across the nodes of the cluster.
More specifically, the Hadoop implementation of the Contrail algorithm in the Map phase scans each read and emits the key-value pairs (u, v) corresponding to overlapping k-mer pairs that form an edge. After the map function completes, a key sorting phase that is executed in parallel groups together edges for the same K-mer based on the key value. The initial graph construction creates a graph with nodes for every K-mer in the read set. This is followed by aggregation of identical K-mers in the Reduce phase, where also linear paths of the de Bruijn graph are calculated and continuously overlapping K-mers are simplified into single graph nodes representing longer stretches of sequence.
Contrail Assembly Steps: The Contrail source code was downloaded from sourceforge.net repository (http://sourceforge.net/apps/mediawiki/contrail-bio/index.php?title=Contrail). Since Contrail uses Hadoop MapReduce programming framework, the reads file from the fish dataset were stored in the Hadoop File System (HDFS), on a local Hadoop cluster with 5TB storage capacity distributed across 8 nodes, with each node having 32GB RAM and 16 cores memory. In order to run the assembly the following steps were followed:
1) Reads files were downloaded from Assemblathon database using wget http://bioshare.bioinformatics.ucdavis.edu/Data/hcbxz0i7kg/Fish/62F6HAAXX.1.1.fastq.gz
2) Copy the unzipped fastq files into the Hadoop File System (HDFS) using ‘put’ command: /opt/hadoop/bin/hadoop/fs –put/source folder/destination folder in HDFS. ‘Put’ command copies files from the local file system to the destination HDFS, also splitting and distributing the file equally across several compute nodes on the cluster.
3) From within the Hadoop Server enter the directory storing the source code for contrail assembler.
4) Run the assembly using the command: contrail.pl –reads [readfile_path] –hadoop [destination folder] –start [stage_of_assembly] [K-mer length].
5) From the output directory obtain the statistic folder, final graph folder and the final contig folder (fasta format) using the Hadoop ‘get’ command. ‘Get’ command copy files to the local file system from HDFS.
6) Extract the files in the final graph folder using cat and gunzip command and store all the graph in a single file: cat part* | gunzip >final_graph.
7) Parse the final graph with the perl script contrail_stats.pl and save the output in a file.
Velvet Assembly Steps: The Velvet assembler was downloaded from the EBI website (http://www.ebi.ac.uk/~zerbino/velvet/). The first component of the assembler is Velveth takes in a number of sequence files, produces a hash table, and then outputs two files in an output directory, “Sequences” and “Roadmaps”, which are necessary for the next step of the assembler called Velvetg. Users need to provide the output directory folder, file format of the sequence stored in the read file, hash length or K-mer length, read type, and read file name. The second Velvetg component of the assembler is where de Brujn graph is built. It has various options like specifying coverage cutoff or minimum length of the contigs to output. Velvet has optional parameters that allow to assemble paired reads data as well. To activate the use of read pairs, two parameters must be specified: the expected (i.e. average) insert length (or at least a rough estimate), and the expected short-read coverage. The insert length is understood to be the length of the sequenced fragment, i.e. it includes the length of the reads themselves.
1) Run Velveth on one fourth of the entire dataset using unpaired reads using the command velveth/outputdir 65 –fastq –short read_file_1 read_file_2.
2) Run Velvetg on the output obtained from Step 1. using velvetg/outputdir.
3) Run the Velvet on paired read files using velveth/outputdir 65 –fastq –shortPaired read_file_1 read_file_2.
4) Run velvetg on the output obtained from Step 3 using velvetg output_directory/-ins_length 101 -exp_cov 20.
5) After the assembly is complete, parse the stats.txt file from the output directory using the Perl program velvet_stats.pl.
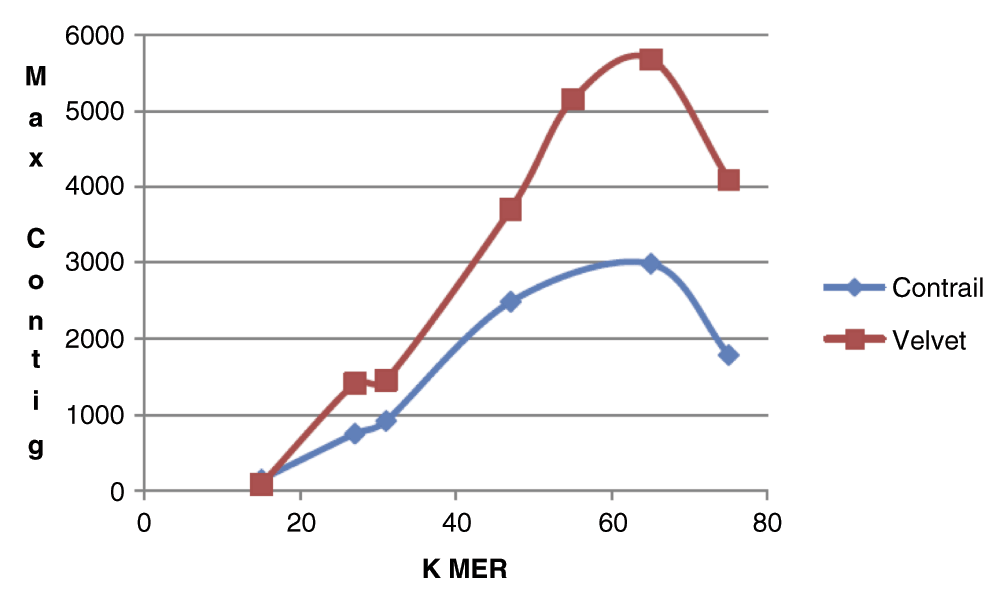
The comparison of the de novo assembly quality for the Velvet and Contrail algorithms presented in the current study, is based on the size of the dataset and assembly statistics using both paired and un-paired sequence data. In the first phase of the research, we compared assembly results for a range of K-mers, using a minimal portion of zebrafish data set (single lane un-paired, approximately 2X coverage). For this dataset, Figure 2 shows the graph plot with the relationship between the K-mer value and the maximum contig size in the assembly output for both Velvet and Contrail assemblers. The graph indicates that the maximum contig length increases with increase in the K-mer size, but above K-mer value of 65 that is a little more than half of the actual read length (101 for our dataset), the contig size decreases indicating lower assembly quality. The reason is that at larger K-mer lengths, the DBG algorithm connects two K-mers with an edge on the assembly graph only if K-1 length suffix of the first K-mer is same as K-1 length prefix of the second K-mer. As the K-mer approaches the actual read size, the probability that any pair of K-mers will have a K-1 length of suffix/prefix identity, decreases significantly given also the presence of sequencing errors. From Figure 2 it is also apparent that for both assemblers the maximum contig size was achieved at K-mer size 65, and therefore the remaining experiments in this study were carried out using that specific value.
In the next step, assembly was performed with Velvet for the same 2X coverage dataset using paired reads, but no significant difference was observed in the maximum contig size (Table 1, row 1–2). Paired read assembly was not performed for Contrail, as the algorithm implementation used in the current study did not provide this feature. The un-paired datasets with Contrail returned half the maximum contig size of Velvet (Table 1, row 3), and took significantly more computing time to complete due to the overhead required for initiating the parallel computation with the Hadoop cluster (see Methods section for details).
In the second phase of the research we attempted to perform assembly using the entire dataset of the zebrafish genome that has approximate coverage of 192X. For running the assembly using Velvet, first the sub-command Velveth was used that creates a “roadmap” of read overlaps, followed subsequently by Velvetg to create and traverse the de Bruijn graph. While we used a 1 Terabyte RAM memory server to run the assembler and the Velveth step of the assembly completed, Velvetg failed to complete, and similar situation was observed when using paired reads (Table 1, row 4–5). The error reported by the Velvet software in both cases indicated that there was not sufficient memory for the completion of the assembly computation. On the other hand, the Contrail assembler successfully completed the run and returned the best N50 score and largest contig size in the current study, albeit requiring 240 hours running time (Table 1, row 6).
Since Velvet failed to complete the assembly for the entire genome, a partial set of the reads from the zebrafish dataset was used as input. First, by using one-fourth of the total dataset the assembly completed in nine and twelve hours for un-paired and paired reads respectively (Table 1, row 7–8). In this case, there was significant difference in the assembly quality between paired and unpaired reads, with the maximum contig size for unpaired reads approximately 7,000bp shorter when compared to paired reads. Next, one-half of the zebrafish dataset was used as input to Velvet, and with unpaired reads the total assembly time completed successfully in 24 hours (Table 1, row 9). In detail, the Velveth step consumed approximately 8hours, while Velvetg required an additional 14 hours. The maximum contig size obtained was about half of that with the Contrail assembler when using the full dataset of unpaired reads (Table 1, row 6). In addition the assembly for Velvet with one-half of the unpaired reads dataset, was found to be comparable to the Velvet results using one-fourth of the paired reads (Table 1, row 8–9). This demonstrates that is feasible to achieve better assembly with less data, when paired end reads are available. Finally, using half of dataset with paired reads, the Velveth step completed in 10hrs while Velvetg failed on our 1TB compute server due to insufficient memory space (Table 1, row 10).
The present study presents an example comparison of performance characteristics for distributed genome assemblers that leverage parallel computing and commodity, cloud computing clusters, versus assemblers that require large memory, specialized and expensive compute servers. The overall conclusion is that for assembling small datasets, reads from bacterial genomes or small eukaryotic genomes, it is better to use assembly software such as Velvet. Nonetheless, for larger genomes and datasets a single server cannot scale, and the assembler fails to load the entire graph into the memory and the assembly does not complete. Genome assemblers such as Contrail present an alternative option, as it performs assembly in parallel using cloud computing and commodity server clusters. Contrail is based on the Hadoop programming framework that is open source, freely available and also can be installed on local cluster. If a local clusters are not available, Hadoop compute servers can be rented from cloud providers. Depending on the size of the assembly to be carried additional nodes can be attached or rented as needed, and expand the capacity of the Hadoop cluster on the cloud or locally. Therefore, additional research by the bioinformatics community on cloud-based, scalable assemblers can result in cost effective solutions for assembly of genomes of any size for both well established institutions or smaller, academic research groups.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)