Keywords
canine, non-Hodgkin, lymphoma, lymphoma, progenitor, cells, ABCB1/P-glycoprotein, valspodar
canine, non-Hodgkin, lymphoma, lymphoma, progenitor, cells, ABCB1/P-glycoprotein, valspodar
The importance of tumor-propagating cells in the pathogenesis of cancer is becoming increasingly well recognized1. However, there are only few reports supporting the existence of such cells in human lymphoma cell lines or in transgenic lymphoma mouse models2–5. Our group identified a subset of lymphoid progenitor cells (LPCs) in primary canine B-cell lymphomas that were characterized by co-expression of hematopoietic progenitor antigens CD34, CD117, and CD133, the B-lymphoid lineage marker CD22, and the common leukocyte antigen CD456. These LPCs had phenotypic properties consistent with tumor-initiating or tumor-propagating cells (TIC/TPC); they also persisted in the xenotransplantation setting, suggesting that they were relevant to the biology of this disease in vivo6. When compared with the bulk of the tumor cells, LPCs showed significantly lower expression of 44 genes across the genome, mapping to cell cycle and transmembrane signaling pathways7. This indicated that LPCs exhibit the characteristic “slow proliferation” seen in normal bone marrow-derived hematopoietic stem cells and in TIC/TPC in other cancers.
One common feature of TIC/TPC in solid tumors is the expression of ATP binding cassette (ABC) transporter proteins such as ABCB1 (multidrug resistance protein-1 or P-glycoprotein) and ABCG2 (breast cancer resistance protein)8. ABC transporter proteins confer drug resistance by actively transporting drugs from the intracellular space to the extracellular space, thereby preventing the interaction of these drugs with their intracellular targets. In the case of ABCB1, expression has been shown to confer resistance to vinca alkaloids, anthracyclines, taxanes, epipodophyllotoxins, and other drugs9,10.
Genome-wide gene expression profiling data showed that mRNAs for ABCB1 and ABCG2 were expressed in several types of spontaneous canine lymphomas, including diffuse large B cell lymphoma (DLBCL) and marginal zone lymphoma (MZL)11. Valspodar (PSC-833) is a selective ABC transporter inhibitor with an acceptable safety profile. Specifically, valspodar had acceptable toxicity when given alone and in combination with cytotoxic chemotherapy in Phase I/II clinical trials in humans with several types of cancer and in one study of dogs with naturally occurring osteosarcoma treated with doxorubicin12–16. These favorable toxicological and pharmacokinetic profiles made valspodar an attractive candidate for targeting LPCs, especially because a safe protocol had been previously established for its neoadjuvant use to inhibit ABCB1 in dogs receiving doxorubicin chemotherapy14. This precedent allowed us to test whether valspodar used in a comparable setting would enhance chemosensitivity of LPCs and extend the time in remission for dogs with spontaneous large B-cell lymphomas.
Clinical grade valspodar (PSC-833) was provided by Novartis Pharma AG (Basel, Switzerland). Valspodar was compounded for use in pet dogs by Custom Rx Compounding Pharmacy (Roy D. Katz R. Ph., Richfield, MN). Capsules containing 100 mg valspodar or placebo (compounding materials without valspodar) were formulated with the same method used to compound cyclosporine-A for oral use in dogs, since these compounds share a high degree of structural similarity. Activity of the compounded valspodar was confirmed using the side population assays described below. Research grade valspodar and verapamil were purchased from Sigma-Aldrich (St. Louis, MO) and were diluted in dimethyl sulfoxide (DMSO; Sigma-Aldrich) for use in vitro. Lymphoma cells were maintained in short-term culture as described6,17,18. COSB hemangiosarcoma cells were maintained as adherent cultures as described19.
This was a double blinded, placebo-controlled trial with 10 dogs in each study arm. The main statistical endpoint was a change in LPCs following treatment. The hypothesis was that a significant reduction in the number of LPCs in blood and/or in lymph node cells would occur in dogs treated with valspodar, but not in dogs receiving the placebo. The sample size of 10 dogs per group was selected to provide 80% power to establish a difference of ± 2 S.D. in LPCs pre-and post-valspodar or placebo treatment within and between groups. The study was not powered to detect significant differences in duration of remission or overall survival. However, outcomes were recorded to evaluate trends that could be used to design future studies. Inclusion criteria included (1) clinical diagnosis of multicentric lymphoma (WHO stage I-V); (2) confirmed WHO classification of large B-cell lymphoma (DLBCL or MZL in transition)20; (3) favorable performance status with an expected survival time of > 30 days; (4) body weight more over 15 kg (to allow adequate blood sampling) and less than 40 kg (to ensure dosing feasibility); (5) platelet count ≥100,000/ml and packed cell volume ≥30%; and (6) informed pet owner consent in writing. Exclusion criteria included (1) disease substage b; (2) any previous therapy for lymphoma, including corticosteroids; (3) lymphomas classified as other than DLBCL or MZL in transition; (4) dogs from herding breeds with high frequency of inactivating MDR-1 polymorphisms21,22; and (5) significant co-morbidities, such as renal or hepatic failure, congestive heart failure, or clinical coagulopathy. There were no restrictions based on age, gender, neuter status, or other physical parameters.
Treatment costs for eligible participants up to $2500 were paid by study funds through the end of the chemotherapy protocol. The study was conducted with approval and under the oversight of the University of Minnesota Institutional Animal Care and Use Committee (IACUC Protocol 1011A92815 “Ablation of tumor initiating cells by P-glycoprotein inhibition: Proof of principle study in canine diffuse large B-cell lymphoma”). The trial design and implementation conformed to the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidelines23 where they apply to studies in companion animals. The flow of participants is provided in Figure 1. The demographic composition of the study population after unblinding is provided in Table 1. The timing of each procedure is shown in Table 2.
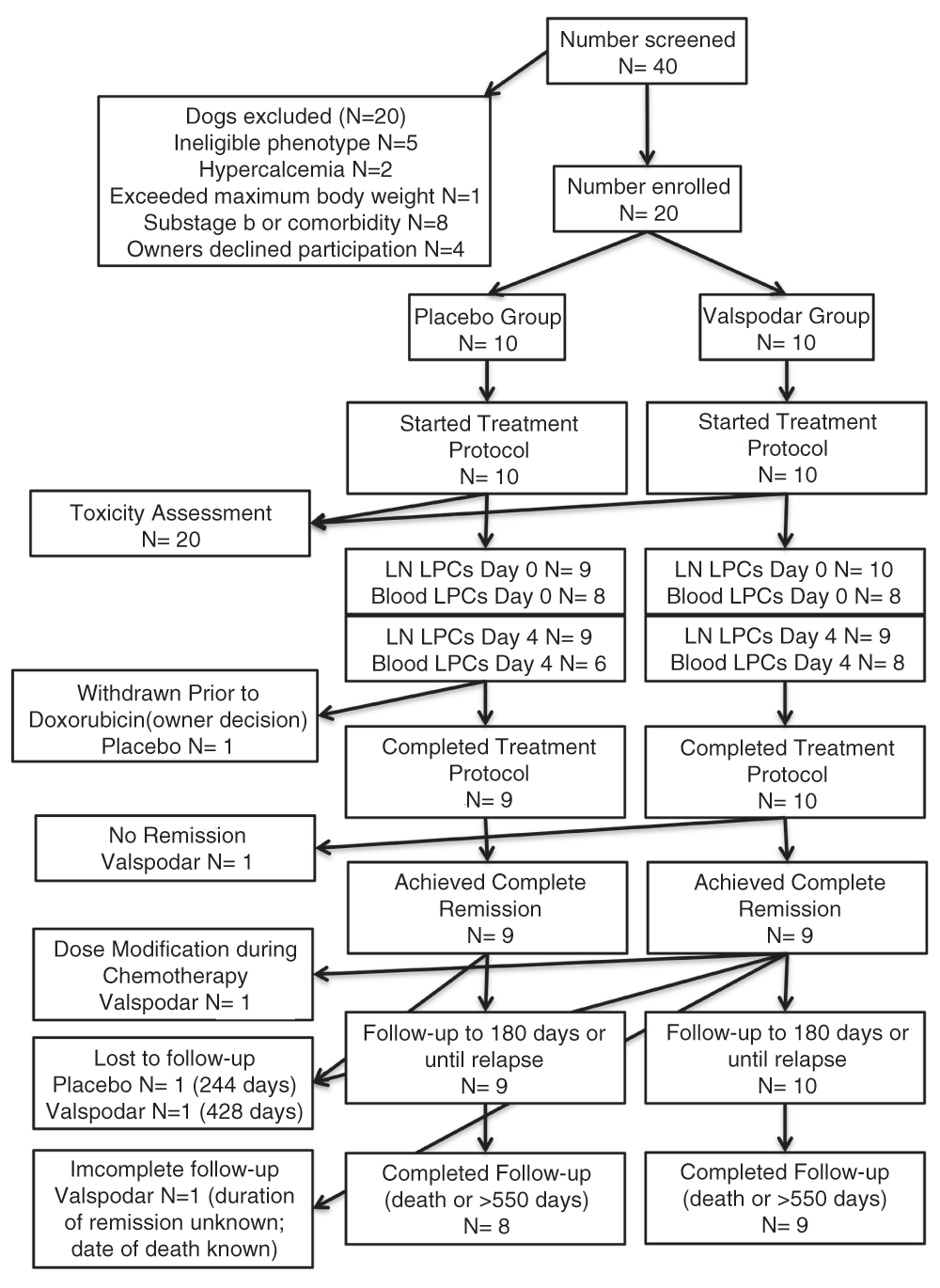
Flow chart with details of dogs enrolled in the study and exclusions from each of the measured endpoints.
Incisional wedge biopsies collected during eligibility screening before treatment (Day 0) and tru-cut biopsies collected on the fourth day of neoadjuvant treatment for enrolled dogs (Day 4) were processed as described24. Briefly, representative sections from each biopsy were fixed in 10% neutral buffered formalin for 24 hours and embedded in paraffin for routine histological analysis. Sample processing, staining, and immunohistochemical stains were done by the Comparative Pathology Shared Resource of the Masonic Cancer Center, University of Minnesota. Samples were classified according to the modified WHO scheme for canine lymphoma based on cell morphology, immunophenotyping using antibodies against human CD3 (AbD Serotec Cat# MCA1477T RRID:AB_10845948), human CD20 (Lab Vision Cat# RB-9013-P0 RRID:AB_149766), and CD79a (clone HM47/A9, Cat# CM 067 C RRID: pending), and available clinical history by two board certified veterinary pathologists (TDO and DMS). The remainder of the biopsy samples was used to prepare single cell suspensions to support the diagnoses through flow cytometry; these suspensions were cryopreserved in liquid nitrogen storage for the following analyses as described6,24.
Blood samples were collected in evacuated EDTA tubes at Day 0, Day 4, and Day 11 to monitor toxicity and to evaluate blood LPCs. Adverse events were recorded and classified according to the Veterinary Cooperative Oncology Group (VCOG) criteria25.
Flow cytometry analysis was performed as described6,17. Briefly, 5 x 105 tumor cells were incubated with dog immunoglobulin G (IgG; Jackson ImmunoResearch, West Grove, PA) to prevent non-specific binding of antibodies to Fc receptors. Cells were stained using fluorescein isothiocyanate (FITC), phycoerythrin (PE), or allophycocyanin (APC) and conjugated antibodies against dog CD3 (clone CA17.2A12, AbD Serotec Cat# MCA1774F RRID:AB_2291174), dog CD4 (clone YKIX302.9, AbD Serotec Cat# MCA1038F RRID:AB_321271), dog CD5 (clone YKIX322.3, AbD Serotec Cat# MCA1037F RRID:AB_322643), dog CD8 (clone YCATE55.9, AbD Serotec Cat# MCA1039PE RRID:AB_322646), dog CD45 (clone YKIX716.13, AbD Serotec Cat# MCA1042F RRID:AB_324047, Cat# MCA1042PE RRID:AB_322644, and AbD Serotec Cat# MCA1042APC RRID:AB_324810), dog CD21 (clone CA2.1D6, AbD Serotec Cat# MCA1781PE RRID:AB_323238), human ABCB1 (clone UIC2, eBioscience Cat# 17-2439-42 RRID:AB_10736477), and human ABCG2 (clone 5D3, eBioscience Cat# 12-8888-82 RRID:AB_466219). Anti-human CD22 antibody (clone RFB4, Abcam Cat# ab23620 RRID:AB_447570) was labeled using the Zenon anti-mouse IgG1 Alexa-Fluor 647 labeling kit (Invitrogen-Molecular Probes, Carlsbad, CA). LPCs were detected by a cocktail of antibodies directed against human CD34 (clone 1H6, BD Biosciences Cat# 559369 RRID:AB_397238), human CD117 (clone YB5.B8, BD Biosciences Cat# 555714 RRID:AB_396058), and mouse CD133 (clone 13A4, eBioscience Cat# 12-1331-80 RRID:AB_465848), where the mix was designated as “Progenitor”6. The antibodies directed against human and mouse antigens have been shown to recognize the canine homologs6,18,26. Cells were gated based on their light scatter properties, and dead cells were excluded using 7-amino-actinomycin D (7-AAD; eBioscience) staining. Flow cytometry was performed using a LSRII cytometer (BD Immunocytometry Systems, San Jose, CA), and results were analyzed using FlowJo software (Tree Star, RRID:nif-0000-30575).
Side populations were measured as described27. Briefly, DyeCycle Violet (DCV) (Life Technologies, Eugene, OR) was added to a final concentration of 10 μM, and 5 × 105 cells were incubated for an additional 60 minutes at 37°C with intermittent mixing. Cells were washed, filtered, and maintained on ice until analysis. To exclude dead cells from analysis, 7-AAD was added to each sample immediately before collection. DCV emission was detected using a BD LSRII flow cytometer (BD Biosciences). Valspodar and verapamil were diluted in DMSO for use in this assay. Equivalent amounts of DMSO were added to control samples, and verapamil was used to determine the side population gates. Data were analyzed using FlowJo software (Tree Star, RRID:nif-0000-30575).
RNA prepared from biopsies obtained at diagnosis (Day 0) and on the fourth day of neoadjuvant treatment for enrolled dogs (Day 4) was quantified and assessed for quality as described11,19. Briefly, total RNA was quantified using a fluorimetric RiboGreen assay and the total RNA integrity was assessed using capillary electrophoresis in the Agilent BioAnalyzer 2100 to generate RNA Integrity Numbers (RIN). Samples passed a QC step if they contained >1 µg with a RIN >8. Next-generation RNA sequencing (RNAseq) was done in 14-paired (pre- and post-treatment) samples and two additional pre-treatment samples as described19. Each sample was sequenced to a targeted depth of ~20 million paired end reads. Base call (.bcl) files for each cycle of sequencing were generated by the Illumina Real Time Analysis (RTA) software. Primary analysis and de-multiplexing were performed using Illumina’s CASAVA software 1.8.2 to verify the quality of the sequence data. The end result of the CASAVA workflow was de-multiplexed into FASTQ files for analysis. Bioanalyzer quality control, RNA labeling, microarray hybridization and reading, and RNASeq were done at the University of Minnesota Genomics Center. Data will be available through the National Center for Bioinformatics (Submitted to Gene Expression Omnibus; GEO).
FASTQ files were mapped to the CanFAM3 genome and the resulting BAM files were summarized to fragments per kilobase of exon per million fragments mapped (FPKM) values using CUFFDIFF. Sequences mapped to 13,952 annotated, named genes. Two-group t-tests were used to determine genes that were differentially expressed between the two groups (i.e., pre- and post-treatment). Expression differences with p-value and false discovery rate (FDR) of less than 0.05 were considered significant.
Eligible dogs were randomized into an experimental treatment group that was given encapsulated valspodar (7.5 mg/kg orally every 12 hours for 5 days) or a control group that was given the equivalent encapsulated placebo over the same schedule. Starting on Day 4, every dog received five doses of doxorubicin 21 days apart using a dosing schedule based on a previous study using valspodar in the neoadjuvant setting with single agent doxorubicin chemotherapy in dogs with osteosarcoma14. The first dose was reduced by 30% from the standard (from 30 mg/m2 to 21 mg/m2) to mitigate potential side effects of ABCB1 inhibition by the neoadjuvant valspodar. If no serious toxic effects of combined doxorubicin/valspodar were observed, subsequent doxorubicin treatments were dosed at 30 mg/m2. If toxic effects were observed, the dose remained at 21 mg/m2 and subsequent dose escalation to 30 mg/m2 only occurred if no serious adverse events were recorded following the previous dose. An overview of the treatment and collection of blood and tissue samples is provided in Table 2. The treatment responses were evaluated based on the VCOG criteria for lymphoma in dogs28. The last treatment was given at 111 days; dogs were examined once more at 180 days, which was near the expected median survival for single agent doxorubicin protocol29, and then released to their attending veterinarian. The status for each dog was ascertained by telephone or electronic mail communication with the attending veterinarians and/or the owners periodically thereafter until a death event was recorded or >500 days had elapsed. Relapse was determined using clinical parameters (generalized lymphadenopathy on physical exam) with conventional testing as needed (routine radiographs or ultrasound imaging, fine needle aspirate). Dogs were considered off-study at relapse and were then eligible to undergo rescue therapy (N=11) or enter other clinical studies (N=4).
Serum samples collected on the fourth day of neoadjuvant treatment (Day 4) were stored at -80˚C until analysis. Valspodar was quantified by liquid chromatography/mass spectroscopy (LC-MS/MS) using a high-performance liquid chromatograph (Agilent 1200 Series, Santa Clara CA) coupled with a TSQ Quantum triple stage quadrupole mass spectrometer (Thermo-Electron, San Jose, CA) as described30.
Descriptive statistics (mean, median, minimum, maximum) were recorded for age, gender, breed, and disease stage; for each variable, differences between groups were determined using Fisher’s exact test. Time to remission, duration of remission, and overall survival were recorded in days starting on the date that the dogs first received a clinical diagnosis. The percentage of LPCs in lymph node samples was calculated based on expression of relevant cell surface markers (CD34/CD117/CD133) as a proportion of live, large, CD22+ B cells6. The ΔLPC was calculated as the ratio of LPCs at Day 4 over LPCs at Day 0. The Mann-Whitney Test (Prism 5, GraphPad Software, Inc., La Jolla, CA) was used to determine significance between LPC numbers in lymph nodes from dogs in the experimental treatment and in the control groups. The associations between variables were determined using the Pearson correlation. Differences between groups in duration of remission and overall survival were determined using Kaplan-Meier probability and log-rank tests.
Valspodar is a potent, selective inhibitor of the ABCB1 efflux transporter12,13. To confirm that the clinical grade compound retained potency after compounding, we examined its effect to inhibit DCV efflux using the flow cytometric side population assay. COSB canine hemangiosarcoma cells contain a subpopulation of cells that shows robust dye efflux in this assay27 (Figure 2, Dataset a). The compounded, clinical grade valspodar was as effective as the research grade valspodar in this assay, eliminating >90% of the side population (i.e., it inhibited dye efflux) at concentrations as low as 30 ng/ml (Figure 2, Dataset a). The effect of valspodar was comparable to that observed in verapamil (Figure 2, Dataset a), which inhibits both ABCB1 and ABCG2 at the 50–100 µM concentrations used in this assay.
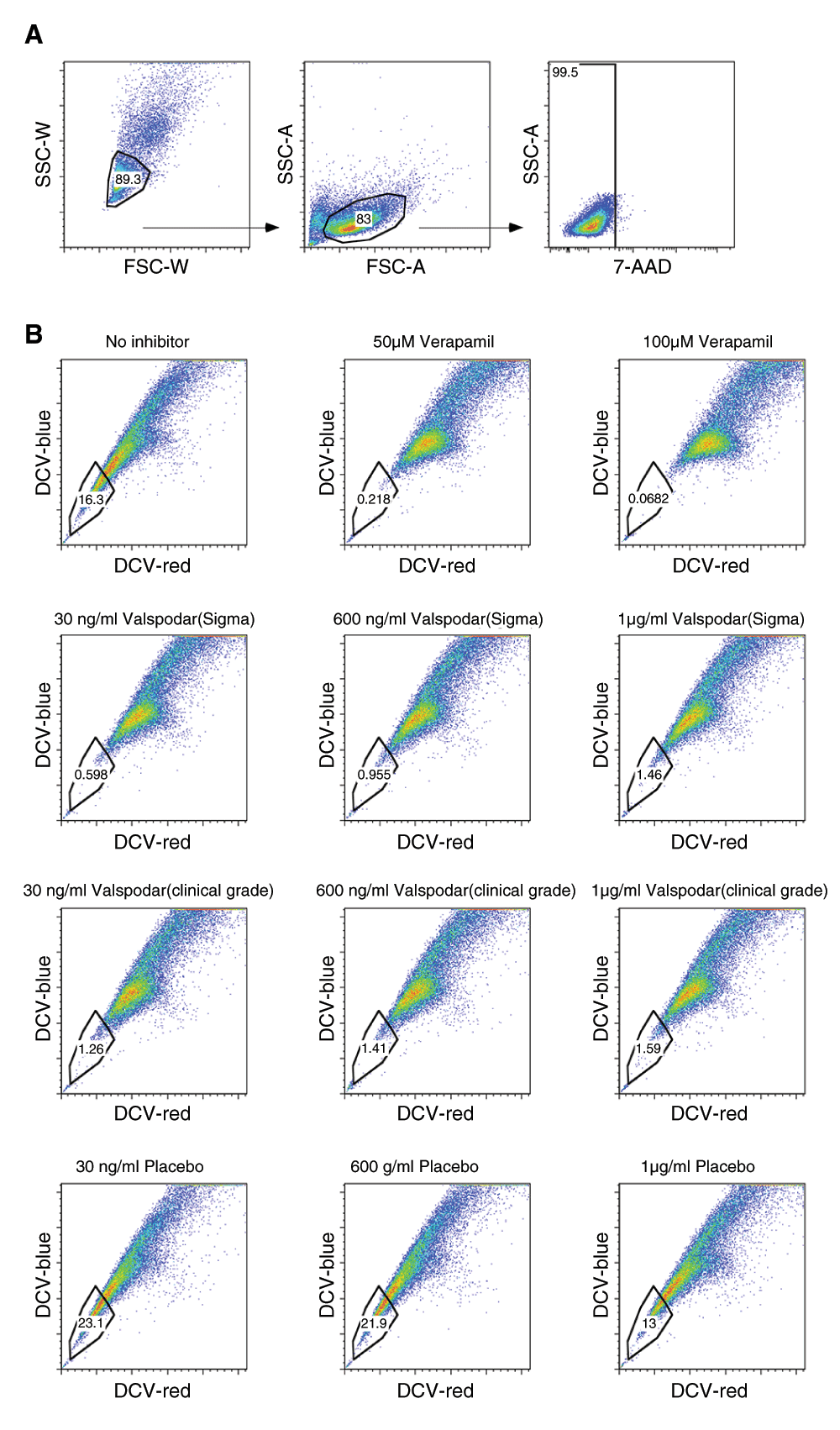
Side population analyses were done as described in Materials and methods using cultured COSB canine hemangiosarcoma cells. (A) Live cells were gated based on light scatter properties and exclusion of 7-AAD, and (B) the side populations were determined based on DyeCycle Violet (DCV) efflux. Verapamil was used to inhibit ABCB1 and ABCG2 at 50-100 µM concentrations. Clinical grade and research grade valspodar was used at concentrations that were achieved in the plasma of dogs in the study (30 – 600 ng/ml) as well as at the saturating dose of 1 µg/ml. The Y-axis is DCV-blue (450+/-50 nm) emission while the X-axis is DCV-red (660 +/- 40 nm) on the LSR-II. Data were analyzed and dot plots were created in FlowJo.
Excluding dogs that had received previous chemotherapy, 40 dogs were screened for eligibility. Twenty dogs were eligible and enrolled in the trial. Of the 20 dogs that were excluded, 5 dogs had a lymphomas that were classified as other than DLBCL or MZL in transition (specifically, three had T-cell lymphoma, one had an indolent type of lymphoma, and one had disease largely confined to spleen with minimal peripheral lymphadenopathy that precluded biopsy) and 15 dogs had hypercalcemia (N=2), lymphoma in substage b or an ongoing co-morbidity (N=8), exceeded the maximum allowable body weight (N=1), or the owners declined participation (N=4).
Of the twenty dogs enrolled, 10 were randomized to each group. The distribution of dogs according to demographic characteristics is shown in Table 1. The composition of the study population was predictable31,32, and there were no statistically significant differences in any category between the experimental treatment group and the control group. One dog in the placebo group did not receive doxorubicin chemotherapy after the neoadjuvant period per its owner’s decision. This dog was censored in the outcome assessments.
Six dogs, including three in the placebo group and three in the experimental (valspodar) group had reportable events during the study (Table 3). The most common toxicities observed in both groups were grade-1 and grade-2 inappetence, lethargy, vomiting, and diarrhea. No grade-4 or grade-5 toxicities were observed, although one event was potentially dose limiting. One dog had grade-2 hematological toxicity (neutropenia and thrombocytopenia) after the first administration of doxorubicin. The doxorubicin dose for the second administration was maintained at 21 mg/m2 and no toxicity was observed. However, the owner only permitted subsequent doxorubicin doses to be escalated to 24 mg/m2. The dog that was withdrawn after neoadjuvant placebo had grade-2 gastrointestinal toxicity and grade-1 lethargy.
| Dog ID Time of event | Placebo Group | Valspodar Group |
|---|---|---|
| MN06 Day 3 | Inappetence (grade 1) | |
| MN08 Day 2 | Inappetence (grade 2) Lethargy (grade 2) | |
| PD02 Day 11 | Gastrointestinal2 (grade 2) Lethargy (grade 1) | |
| PD05 Day 1 after first dose of doxorubicin3 | Lethargy (grade 1) Gastrointestinal4 (grade 2) Hematological5 (grade 2) | |
| PENN02 Day 2 and Day 5 Days 6–11 after first dose of doxorubicin | Gastrointestinal6 (grade 1) Inappetence (grade 3) Lethargy (grade 2) | |
| PENN05 Day 4 Day 11 | Bilateral scleral congestion Suspected hyphema OS (grade 2) Lymphadenopathy Uveitis OD (grade 2) |
1Owner elected to withdraw dog from study prior to receiving doxorubicin
2Vomiting and diarrhea
3Dog’s second doxorubicin treatment was dosed at 21 mg/m2; similar toxic effects were not observed. However, the dog’s owner only permitted subsequent doxorubicin doses to be escalated to 24 mg/m2
4Diarrhea
5Neutropenia (grade 2) and thrombocytopenia (grade 1)
6Vomiting
Blood and lymph node LPCs were quantified for each dog at diagnosis (Day 0) and on the fourth day of neoadjuvant treatment (Day 4) as described in Materials and Methods. Table 4 shows that LPCs were detectable in every sample at a comparable frequency to what was previously reported6. The distribution of lymph node LPCs at diagnosis was narrower in the dogs that received valspodar than in the control dogs (Figure 3A), but the two groups were not significantly different, and neither group showed a statistically significant reduction in LPCs on the fourth day of the neoadjuvant period (ΔLPC). Similar results were observed for blood LPCs, with the exception that the variance in frequency of these cells in blood was noticeably increased on the fourth day of the neoadjuvant period (Figure 3B).
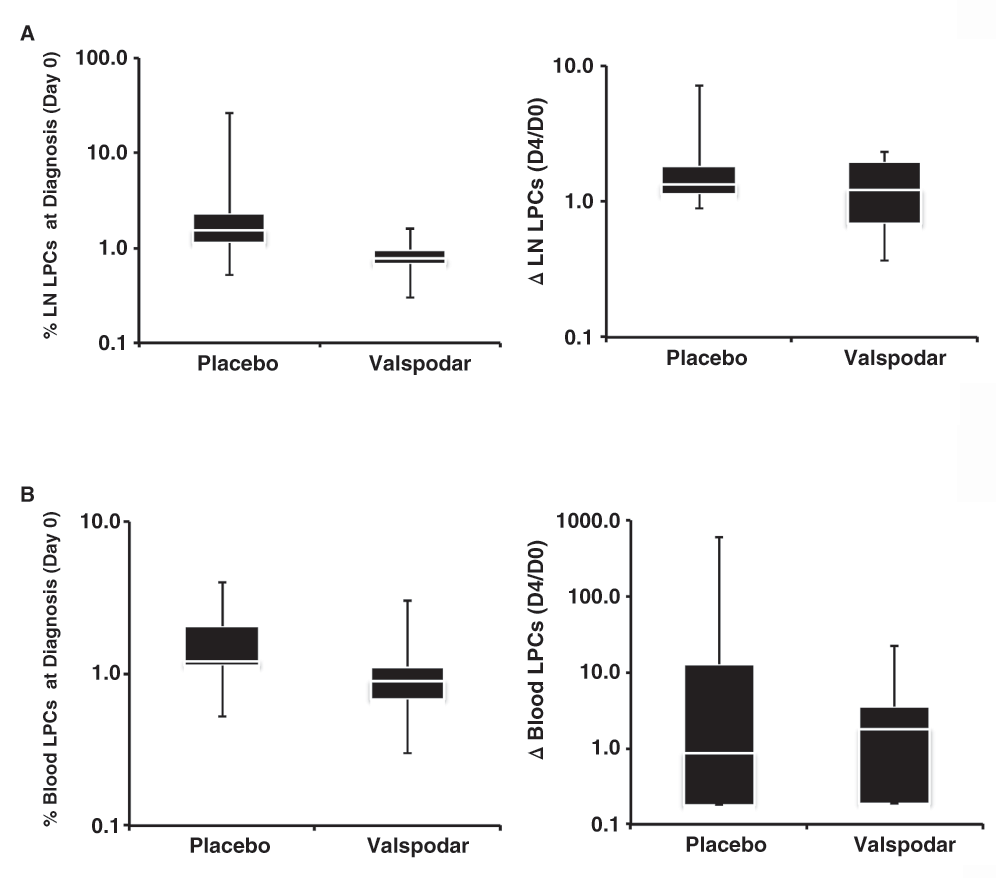
(A) Box plots showing median (white line), 75% confidence intervals, and outliers of the percent LPCs in lymph nodes at diagnosis (top) and relative change in LPCs (bottom) from the time of diagnosis (Day 0) to the fourth day of the neoadjuvant period (Day 4) ΔLPC = 1.0 means no change in the percent in each group of dogs. A LPCs measured at both time points. (B) Box plots showing median (white line), 75% confidence intervals, and outliers of the percent LPCs in peripheral blood at diagnosis (top) and relative change in LPCs (bottom) from the time of diagnosis (Day 0) to the fourth day of the neoadjuvant period (Day 4) in each group of dogs. Data were analyzed and graphs were assembled using MS Excel.
The absence of a treatment effect on total LPCs suggested that we could not reject the null hypothesis that neoadjuvant valspodar did not enhance chemosensitivity of LPCs, and could reflect variable expression of ABC transporters by these cells. Samples from 15 dogs in the study (six in the placebo group and nine in the valspodar group) had sufficient material for analysis of ABCB1 and ABCG2 expression in LPCs at diagnosis. The proportion of ABCB1+ LPCs and ABCG2+ LPCs was variable. In the placebo group, between 1.6% and 52.4% of lymph node LPCs expressed these proteins at the time of diagnosis; in the valspodar group, the range of ABCB1 and ABCG2 transporter expression in lymph node LPCs at the time of diagnosis was 10.0% to 72.7% (Table 5). When we examined the proportion of ABCB1+ LPCs and ABCG2+ LPCs in dogs from each treatment group, we saw an intriguing reversal in the trends with regard to event-free survival (Figure 4), although neither group showed a significant correlation between the number of ABCB1+ or ABCG2+ cells at diagnosis and survival (all the R2 values were less than or equal to 0.42).
| Dog ID | Group | % ABCB1+ LPCs | % ABCG2+ LPCs |
|---|---|---|---|
| MN05 Day 0 Day 4 | Placebo | 43.4 36.6 | 52.4 22.1 |
| MN09 Day 0 Day 4 | Placebo | 1.7 1.1 | 1.6 0.8 |
| MN02 Day 0 Day 4 | Valspodar | 60.8 47.9 | 68.8 61.3 |
| MN10 Day 0 Day 4 | Valspodar | 55.7 33.5 | 53.4 49.6 |
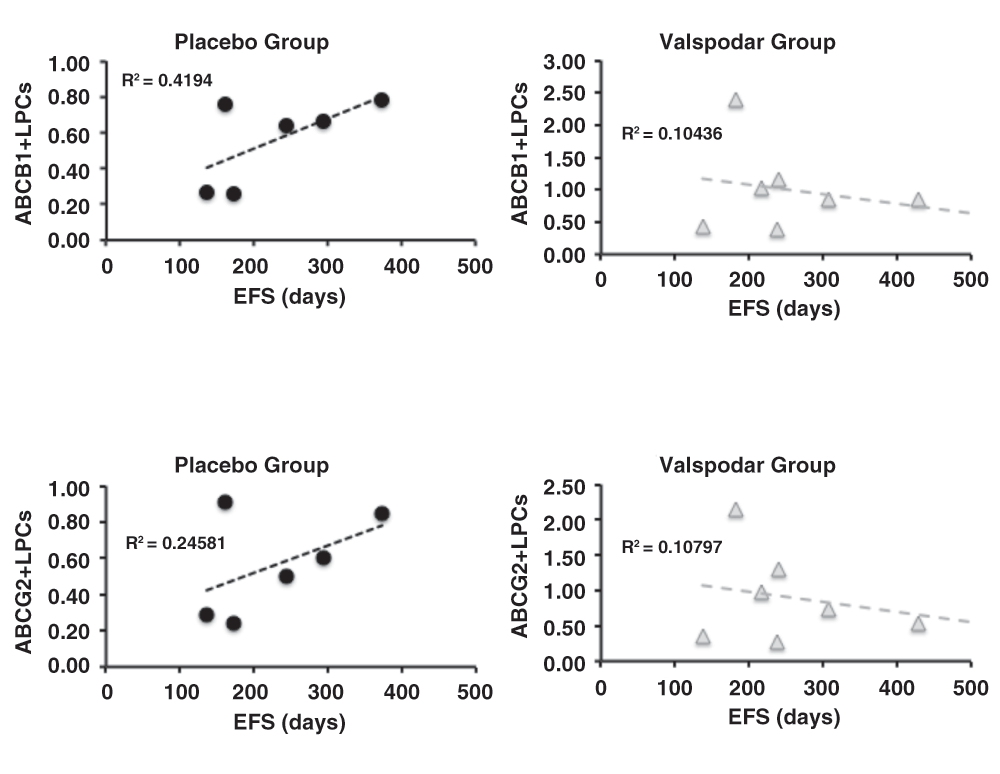
Dot plots showing the relationship between ABCB1 expression and event-free survival (EFS) in days (top) and between ABCG2 expression and EFS in days (bottom) in dogs treated with placebo (N = 9) or with neoadjuvant valspodar (N = 9) where samples were available for these measurements. The dashed lines represent linear regressions and their R2 values are indicated on each graph. The Y-axis represents the % of ABC+/Progenitor+lymph node B cells. Data were analyzed and graphs were assembled using MS Excel.
Samples from four dogs (two in the placebo group and two in the valspodar group) had sufficient material for analysis of ABCB1 and ABCG2 to determine if valspodar specifically reduced the number of ABCB1+ and ABCG2+ LPCs in paired pre-and post-treatment samples. There was a quantifiable decrease in the frequency of ABCB1+ and ABCG2+ LPCs, but this change was comparable between the two dogs that received valspodar and the two dogs that received placebo (Table 5 and Supplementary Figures 1A–1D, Dataset b).
We examined if the inhibition of ABCB1 activity with valspodar changed genome-wide patterns of gene expression in lymph nodes from dogs in both groups. Paired pre- (Day 0) and post- (Day 4) treatment samples were available from five dogs in the placebo group and from nine dogs in the valspodar group. One additional pre-treatment sample from dogs in each group was available and included in the analysis, making a total of 16 pre-treatment samples and 14 post-treatment samples. We did not identify any genes with significantly differently expression between groups or between pre- and post-treatment samples in the placebo or the valspodar groups.
The observation that valspodar treatment did not specifically alter the total blood or lymph node LPCs or the frequency of ABCB1+ and ABCG2+ LPCs, and that it did not lead to significant changes in gene expression of lymph node cells, could be attributed to poor bioavailability. To evaluate this possibility, we examined the purity of the compounded, encapsulated drug and the levels of valspodar in serum samples obtained at Day 4 from seven dogs using LC-MS/MS. Valspodar was undetectable in placebo capsules, and the purity of the compounded capsules was 104% as compared to research grade valspodar.
Valspodar was also undetectable (<5 ng/ml) in dogs that received placebo, but it was present at detectable levels in each of four dogs that received compounded valspodar capsules (34, 63, 375, and 623 ng/ml, respectively). This is equivalent to levels between 0.025 to 0.5 µM on the fourth day of twice-daily administration, which is in the range seen in dogs where valspodar was given at the same dose in an oil-based drinking solution14.
Eighteen treated dogs achieved clinical remission, defined as a complete response (disappearance of all evidence of disease with all lymph nodes shrinking to non-pathologic size within the judgment of the evaluator) after the first dose of doxorubicin. One dog in the valspodar group did not achieve clinical remission, but survived with stable disease for 428 days. One dog in the placebo group never received doxorubicin and was censored from this analysis. This dog was treated with palliative intent using prednisone only; it failed to achieve remission and died 59 days after diagnosis.
The time to remission after the initial valspodar treatment ranged from 7 to 106 days (after doxorubicin) in the placebo group, and from 7 to 105 days (after doxorubicin) in the valspodar group (excluding the dog that never achieved remission). There were no differences between groups with reference to the median time to remission, the median (or range) duration of remission, the number of dogs alive at the 180-day milestone, or the number of dogs alive at 500 days (Table 6). The event-free survival and overall survival times for each group are shown in Figure 5.
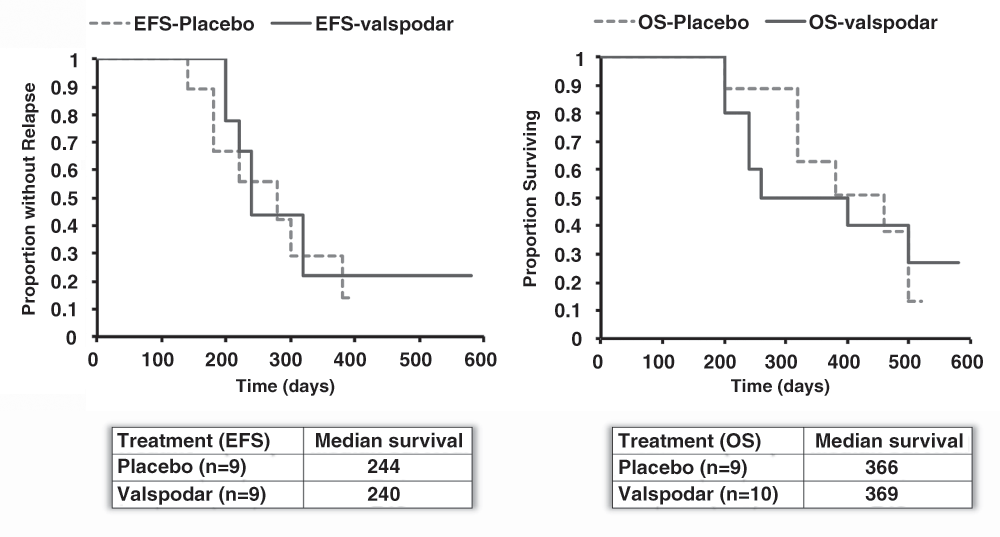
Kaplan–Meier analysis of event-free survival (top) and overall survival (bottom) in dogs treated with doxorubicin with the addition of neoadjuvant placebo or valspodar. The table below the graphs shows the median event-free and overall survival for each group. Data were analyzed and graphs were assembled using MS Excel.
To test the hypothesis that LPCs contribute to disease progression, we examined if there were direct or inverse correlations between the proportion of LPCs at diagnosis and the ΔLPCs with duration of remission as well as with overall survival for dogs in the valspodar and control groups, individually and for all of the dogs in the study. Figure 6A and 6B show scatterplots illustrating no correlations between the proportion of lymph node LPCs at diagnosis and the ΔLPCs (D4/D0), respectively, and event-free survival (duration of remission) and overall survival. The results were similar when we analyzed correlations between the proportion of blood LPCs at diagnosis or ΔLPCs and survival outcomes (data not shown).
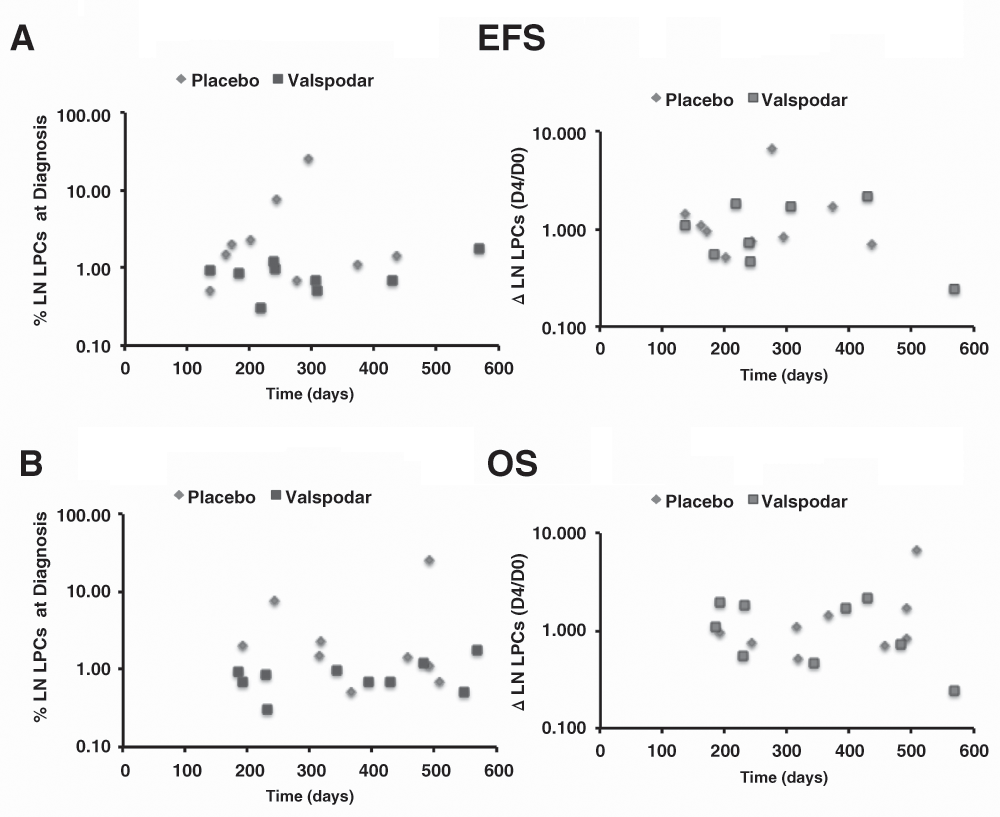
(A) Dot plots showing the relationship between the percent of lymph node LPCs at diagnosis and EFS (N=9), and the relative change in LPCs from the time of diagnosis (Day 0) to the fourth day of the neoadjuvant period (Day 4) and EFS (N=8), in days in dogs treated with placebo (N = 9) or with neoadjuvant valspodar. (B) Dot plots showing the same relationships for overall survival (OS, N=9 and N=10 for LPCs at diagnosis and for ΔLPCs, respectively). Data were analyzed and graphs were assembled using MS Excel.
We conducted a double-blinded, placebo controlled study in 20 dogs to determine whether valspodar used in the neoadjuvant setting would sensitize LPCs to doxorubicin and increase the length of remission in dogs with therapy naïve large B-cell lymphoma. Our results confirmed the previous observation from Cagliero et al.14 showing that valspodar can be safely administered to dogs twice daily at a dose of 7.5 mg/kg. Furthermore, we verified that CD22+/CD34+/CD117+/CD133+ LPCs constitute between 0.3 – 2% of lymph node B cells and 0.001 – 3% of peripheral blood B cells in dogs with large cell B-cell lymphomas. The observation that these cells are virtually undetectable in lymph node samples from healthy dogs, while they exist in a steady state in canine B-cell lymphomas even in the xenotransplantation setting6, suggests that they contribute to the maintenance or propagation of the tumor population.
Upregulation of ABC transporters is a well-described mechanism of acquired drug resistance in lymphoma and other cancers, making these proteins attractive targets for pharmacologic modulation33,34. These proteins are transport channels that extrude a variety of compounds, including xenobiotics, from cells. Cells expressing these proteins have been defined functionally as “side populations” based on their ability to exclude fluorescent dyes in flow cytometric assays. The possibility that increased expression of ABCB1 and other transporters was due to selection of cells intrinsically possessing this trait, as opposed to through de novo induction of expression, was proposed more than 20 years ago35 and recapitulated most recently in canine lymphomas in vitro through drug selection, with expansion of a valspodar-sensitive subclone that had increased expression of ABCB1 and ABCG236.
“Side populations” are routinely detectable in canine lymphomas37. In that study, 0.1 to 4% of cells in the canine B-cell lymphoma cell lines GL-1 and 17-71 excluded Hoechst 33342 and expressed detectable levels of ABCB1 and ABCB2. A dye-excluding side population was also variably detectable in five primary lymphomas. GL-1 cells and one of the lymphoma samples expressed a form of ABCB1 with slower electrophoretic mobility, possibly representing the active, phosphorylated form of this transporter38. ABCG2 was expressed ubiquitously in GL-1 cells and in the five primary lymphomas. However, the side population identified by Kim and colleagues was insensitive to verapamil and to fumitremorgin-C37, suggesting that the dye exclusion activity might have been mediated by an ABC transporter distinct from ABCB1 and ABCG2.
The notion that cells expressing ABC transporters can behave like cancer stem cells in lymphomas is not universally accepted. Indeed, the existence of tumor-initiating or tumor-propagating cells (TIC/TPC) or of a hierarchical organization in lymphoid malignancies at all remains a matter of debate39. In acute lymphoblastic leukemias (ALL), models for cells of origin have been proposed, including common hematopoietic progenitors, common lymphoid progenitors, and committed B-lymphoid cells, depending largely upon the molecular subtype of ALL. In preliminary experiments, samples from two human patients with ALL included a subset of CD117+ cells that were present at a similar frequency to LPCs in canine lymphoma (D. Ito and J. Modiano, unpublished results); however, the functional significance of this finding remains to be determined. The evidence for TIC/TPC in solid lymphomas is even more sparse. Drug resistant TIC/TPCs were defined in follicular lymphoma using side population assays and increased expression of ABCG22. Tumor formation in these cells was limited by an obligate interaction with follicular dendritic cells in the microenvironment niche, which was mediated through the CXCR4 chemokine receptor. TIC/TPC were similarly identified using side population assays in a mouse model of mantle cell lymphoma4, and more recently in human anaplastic lymphoma kinase (ALK)-positive and -negative anaplastic large cell lymphomas40.
Next generation sequencing and genome-wide epigenomic analyses of human DLBCL have revealed a potential mechanism to explain how lymphoid cells might acquire TIC/TPC properties and how this acquisition could be related to the expression of ABC transporters. The gene encoding the enhancer of zeste homolog 2 (EZH2) had gain of function mutations in 7/49 (14%) DLBCL patients sequenced41. EZH2 is a histone methyltransferase that functions as part of the polycomb group complex, which controls the balance between self-renewal and differentiation42. In germinal center (GC) B cells, EZH2 appears to suppress differentiation genes and favor behavior that resembles stem cells43. As in GC DLBCL cells, depletion of EZH2 in Bel/Fu hepatocellular carcinoma cells inhibited proliferation, but in Bel/Fu cells this depletion also increased methylation at the ABCB1 gene, reduced ABCB1 gene and protein expression44, and showed consequent sensitization of these cells to the cytotoxic effects of 5-fluorouracil45. Together, these findings provide a strong rationale for use of neoadjuvant therapies to sensitize TIC/TPCs in lymphoma using ABC transporter inhibitors, at least in a subset of GC DLBCL.
Our data show that LPCs in canine large B-cell lymphoma were heterogeneous regarding expression of ABCB1 and ABCG2, with slightly fewer present in the dogs randomized to the placebo group. Such heterogeneity is consistent with previous observations in human lymphoma samples3. The apparent reversal in outcome trends between the placebo and valspodar groups as a function of the percent lymph node B-cell LPCs at diagnosis was intriguing, and while tempered by the small sample size, it suggests this approach merits additional investigation.
The proportion of ABCB1+ and ABCG2+ LPCs appeared to decrease in the samples from four dogs during the neoadjuvant period where we could perform the analysis; however, the change was unrelated to valspodar, since a reduction of similar magnitude occurred in the dogs assigned to both the placebo and the valspodar groups. Furthermore, statistically significant differences were not found in either the total number of LPCs or in the duration of remission (or overall survival) between groups of dogs treated with valspodar and placebo.
It is worth noting that the duration of remission and the overall survival of dogs in this study slightly exceeded the expectations based on previously published results using single agent doxorubicin29. This could be attributed to improved management of cancer patients over time, but it also could be due to recruitment of a relatively uniform population of dogs based on clinical and pathologic criteria20. The latter possibility highlights the benefits of study designs that narrow disease heterogeneity, particularly for canine lymphoma where each disease entity in this complex is considered as an individual disease.
There are several possible explanations for the absence of clinical improvement in dogs receiving valspodar vs. placebo. First, it is possible that this treatment would be most effective against a specific subset of DLBCL, such as EZH2-mutated GC DLBCL. It has been challenging to separate canine DLBCLs into activated B-cell (ABC) type and GC-type DLBCL11,46, although one study suggested canine DLBCL might be more similar to human ABC type DLBCL47. Second, it must be noted that the study was designed to address chemosensitization of LPCs by valspodar, and the sample size was not powered to reveal if this protocol would significantly improve survival outcomes. Based on our results, we estimate that a clinical trial where we could detect a doubling of the median overall survival (from 12 months to 24 months) in dogs receiving neoadjuvant valspodar would require 35 dogs each in the treatment and in the placebo arms.
Nonetheless, we confirmed absorption and bioavailability of the drug on the fourth day of administration, and we showed that the drug was able to fully inhibit ABC transporter activity in a side population assay even at the lowest dose detected. However, the levels of valspodar required for sustained, active inhibition of ABC transporter activity in vivo have not been conclusively established. For example, when valspodar (50 mg/kg) and paclitaxel (10 mg/kg) were administered concurrently to mice through the oral route, they passed rapidly through the stomach and reached the intestine together, but showed enhanced uptake and plasma levels for paclitaxel48. In rats, oral valspodar was absorbed rapidly and had excellent bioavailability with low hepatic extraction49. In human patients with chemotherapy resistant multiple myeloma, a dose escalation study showed similar pharmacokinetic properties. Orally administered valspodar combined with doxorubicin, vincristine and dexamethasone led to a doubling of area under the curve for doxorubicin levels in the plasma and reduced its clearance by half16. The concentration of valspodar in serum increased proportionately with a dose of up to 15 mg/kg/day, although it reached a maximum effectiveness level vis-à-vis increasing plasma doxorubicin at 5 mg/kg/day where the median trough and peak levels (of valspodar) were 461 ng/ml and 1134 ng/ml, respectively. The treatment regimen was associated with increased toxicity and required dose reduction in more than 50% of the patients (13/22). Yet, 14 of the patients treated had either a partial response or stable disease, and ABCB1 expression in bone marrow plasma cells was reduced in four of the five responding patients examined.
In another study, valspodar was administered concurrently with doxorubicin to 31 cancer patients using an intravenous loading dose of 1–2 mg/kg and a continuous dose of 1–10 mg/kg over 24 hours. Doxorubicin was given immediately at the end of the loading dose and the treatment was repeated every 21 days until there was disease progression or unacceptable toxicity15. As noted in the Sonneveld study16, patients receiving valspodar showed a significantly increased area under the curve for doxorubicin, with a 50% shortening of doxorubicin clearance as compared to controls. The steady-state concentrations of valspodar over the time of continuous administration ranged from 190 ng/ml to 1383 ng/ml with unchanged rates of clearance, and serum from treated patients contained sufficiently high levels of valspodar to inhibit ABCB1 activity in an in vitro bioassay. Dose limiting toxicities were observed only in patients treated with the highest dose of valspodar (2 mg/kg loading dose and 10 mg/kg continuous dose) and 50 mg/kg doxorubicin. One patient (ovarian cancer) had a partial response, but none of the patients in this trial had non-Hodgkin lymphoma15.
The effective serum concentrations and positive bioassay results in these studies are in contrast to those in another series of experiments showing that the concentration required to inhibit ABC transporter activity in vitro under complete serum conditions (cells cultured in 100% fetal bovine serum) is almost a full order of magnitude (8–9 times) higher than the plasma concentrations achieved in clinical trials, probably due to binding of valspodar by serum lipoproteins50. Among the compounds examined, daunorubicin was the most relevant. In 100% serum, the half maximal concentration of valspodar required to inhibit ABCB1-mediated daunorubicin transport was approximately 1.5 µM (or approximately 1800 ng/ml), which is close to the peak levels achievable using continuous infusions15 and almost 3-fold higher than the levels we measured in our study.
It also is possible that inhibiting ABCB1 and ABCG2 in LPCs is insufficient to ablate the population. In our study, 30% to 90% of lymph node LPCs did not express ABCB1 or ABCG2. In addition, the variable sensitivity to verapamil and other ABC transporter inhibitors by LPCs and side population cells in leukemia and lymphoma suggests that these cells might rely on alternative mechanisms of drug export and/or drug resistance. Still, it has been shown that clinically relevant anti-lymphoma immunotherapies including rituximab51 and anti-CD19 antibodies52 induce ABCB1 to translocate out of lipid rafts, reducing its ability to extrude chemotherapy agents such as vincristine and doxorubicin and increasing the chemosensitivity of drug-resistant lymphoma cell lines. We propose that the totality of data continues to support the rationale for implementing treatment approaches for non-Hodgkin lymphoma that target ABCB1 and ABCG2 in the neoadjuvant or the adjuvant settings. These treatments might be most effective for patients with tumors that do not respond to other targeted agents, such as those diagnosed with EZH2-mutant GC DLBCL. Thus, additional work and diligently crafted clinical trials, as well as creative animal models of induced and spontaneous disease, will be needed to establish the significance of LPCs in the pathogenesis of lymphoid malignancies and the potential to improve patient outcomes by targeting the ABC transporter-enriched and the ABC transporter-deficient subsets of these cell populations.
F1000Research: Dataset 1. Data of pilot study on valspodar in neoadjuvant settings for canine B-cell lymphoma, 10.5256/f1000research.6055.d4289753
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)