Keywords
ontology, medical subject headings, MySQL, annotation, phentypes
ontology, medical subject headings, MySQL, annotation, phentypes
Understanding molecular mechanisms underlying both normal cellular processes and disease-causing gene perturbations has numerous applications in clinical diagnostics, personal genomics and engineering1–5. Most of the genomic studies address two major questions: (i) What genomic and molecular markers are associated with an observed phenotype? (ii) What molecular mechanisms lead to that phenotype in the studied organism? Answering these questions and uncovering gene-phenotype relationships mostly relies on experimental research that has already generated very large amounts of high-throughput data stored in public databases6–10. New knowledge about genes and their functions is acquired all the time based on a constant gathering of genomic data. To date there are more than 1500 databases hosting various types of genomic and molecular biology data11 acompanied by increasing number of research publications analyzing newly-generated data12. For this reason integrative algorithms to analyze high-throughput data by mining genomic databases and literature are in the focus of intensive research resulting in many publicly available bioinformatics tools for biologists and clinical researchers6,13–19.
Biologists analyze lists of genes to dissect individual or collective gene involvement in the biological function that is being investigated, for example:
functions of differentially expressed genes identified in a microarray or RNA-Seq experiment;
relationships between a biological process of interest and target genes regulated by a transcription factor identified by ChIP-Seq experiment;
causative relationships between functions of genes found in a chromosomal deletion or duplication identified in a patient and a clinical phenotype of the patient;
identifying candidate genes from gene lists in literature and databases.
Finding meaningful relationships between genes in a large list and a phenotype by manually reviewing the literature and genomic databases is very laborious and time-consuming. Efforts to automate this process mostly have been directed towards the prioritization of human disease genes20,21 and less for model organisms and general phenotypes10. Gene prioritization tools, that can be used to infer relationships between genes and phenotypes, differ from each other with respect to computational algorithms and data sources used in prioritization21–23. In computations, a definition of a phenotype will determine the rules by which the algorithm will mine available data resources to retrieve gene-phenotype links.
The definition of a phenotype widely accepted in biology is “the observable trait or the collection of traits of an organism resulting from the interaction of the genetic makeup of the organism and the environment” meaning different things in different contexts24,25. In medicine the phenotype often refers to disease or abnormality26. In cellular contexts measurable cellular phenotypes are represented by features of cells such as the morphology (shape, size), the behavior (motility, growth), the developmental stage, the expression of specific genes and the rate of bio-chemical reactions8,27.
Specific vocabularies of phenotype terminology are implemented as ontologies containing concepts, the relationships between the concepts and the definitions of both28,29. Specialized phenotype vocabularies are available for model organisms30,31, life sciences32–36 and human diseases37,38. Phenotype can also be defined as a subset of genes known to be functionally related to the phenotype of interest, usually used in gene prioritization algorithms23,39. However, if the phenotype of interest hasn’t been well studied and does not have genes linked to it, then it is difficult or even impossible to use this approach.
Terms of Medical Subject Headings (MeSH) vocabulary can serve as appropriate phenotypic descriptions38. MeSH terms are curated and are assigned to the articles in PubMed to adequately reflect the content of each article since they are meaningfully associated with the biological processes that they denote. Phenotypes in Mammalian Phenotype Ontology (MPO) used in Mouse Genome Informatics (MGI) database40 are also mapped to MeSH terms.
Gene prioritization tools have to establish links between genes and phenotypes by use of some algorithm. Several overlapping categories of tools can be distinguished based on how a phenotype is defined: by training genes known to be related to the phenotype; by keywords and tools designed to work with disease phenotypes. Table 1 lists maintained prioritization tools from these categories that are frequently cited in GoogleScholar. Algorithms defining phenotypes by training genes in prioritization evaluate similarity between training genes and candidate genes. Supervised machine learning (most often kernel methods) are used in this category of tools39,41. Algorithms describing phenotypes by keywords usually use frequencies of gene-associated documents that have keyword matches. Majority of algorithms are designed to prioritize genes with respect to disease phenotypes defined by either the keywords or the training genes or by both.
| Tools | Web Link |
|---|---|
| Endeavour39,41,42 G2D43 ToppGene44,45 GeneWanderer46 MimMiner47 PolySearch48 SUSPECTS49 PhenoPred50 CANDID51 PosMed52 GeneProspector53 | www.esat.kuleuven.be/endeavour/ www.ogic.ca/projects/g2d_2/ toppgene.cchmc.org/ compbio.charite.de/genewanderer/ www.cmbi.ru.nl/MimMiner/ wishart.biology.ualberta.ca/polysearch/ www.genetics.med.ed.ac.uk/suspects/ www.phenopred.org dsgweb.wustl.edu/hutz/ omicspace.riken.jp www.hugenavigator.net |
Endeavour. In Endeavour the phenotype is defined by the training genes. It builds a phenotype model using different sources of genomic information derived from the training genes. Endeavour data sources consist of gene annotations, gene sequences, expressed sequence tags over multiple conditions, protein-protein interaction data and known transcription factor binding sites. The program works with the genes of human, mouse, rat, fly and worm organisms. It builds the model of the phenotype using information of the training genes in each of the genomic sources. It ranks the candidate genes according to how well they compare with the built model. Individual rankings in the Endeavour are combined by the order statistics42. In the table of the ranked genes the explanations are provided about the genes.
ToppGene. The candidate gene prioritization is one of the functions provided by the ToppGene tool. The user submits a set of training genes and a set of the test genes. The ToppGene first finds the significantly enriched annotations for the training genes in multiple data sources: GO annotations, literature, Interaction, Pathway, human and mouse phenotype data, TF binding sites, Cytobands, Co-expression Atlas, Drugs, microRNA and more. The candidate genes are ranked by the similarity of their functional annotations to the enriched annotations in the training genes. The similarity is computed as fuzzy-based measure54 or Pearson correlation coefficient. The user can examine the genes and the enriched terms of the the training set.
GeneWanderer. In GeneWanderer the candidate genes are retrieved from the genomic region given the genomic coordinates. The phenotype is defined either by the disease keyword or by the list of the training genes known to be related to the phenotype. If the phenotype is defined by the keyword then the known genes associated with it are retrieved. The tool measures the distance between the candidate genes and the training genes in the protein-protein interaction network. The tool is specific to the human diseases.
PolySearch. PolySearch allows queries in the form of: Given X find all Y. X and Y can be diseases, tissues, cell compartments, gene/protein names, SNPs, mutations, drugs and metabolites. If the phenotype is defined by keywords, then PolySearch retrieves the documents matching all keywords in the Pubmed, OMIM, DrugBank, Swiss-Prot, Human Mutation Database, Genetic Association Database and Human Protein Database. The ranked list of requested biomedical entities that are associated with the text of the query is returned. The score of the entity is proportional to the number of document matches in the databases. User can browse the results and examine the matching publications and sentences.
PosMed. Positional PubMed is the semantic engine that ranks biomedical entities by the statistical significance of the associations with the provided keywords. The strength of the associations between biomedical entities and keywords is based on the number of the documents they share. The document categories comprise PubMed (PubMed titles, abstracts and MeSH terms), REACTOME (Pathway information from REACTOME), Protein-protein interaction (Protein-Protein Interactions in Human and Mouse from IntAct and Arabidopsis from AtPID), Gene ontology Human disease ontology Mammalian phenotype ontology Microarray based co-expression data for Arabidopsis. Given the keyword defining the phenotype and the type of biomedical entity to score (either gene or metabolite or drug) the PosMed returns list of the scored entities linked to the phenotype, sorted according to the strength of the connection between them. The PosMed supports human, mouse, rat, arabidopsis and rice organisms. The user can browse through all documents of the established links.
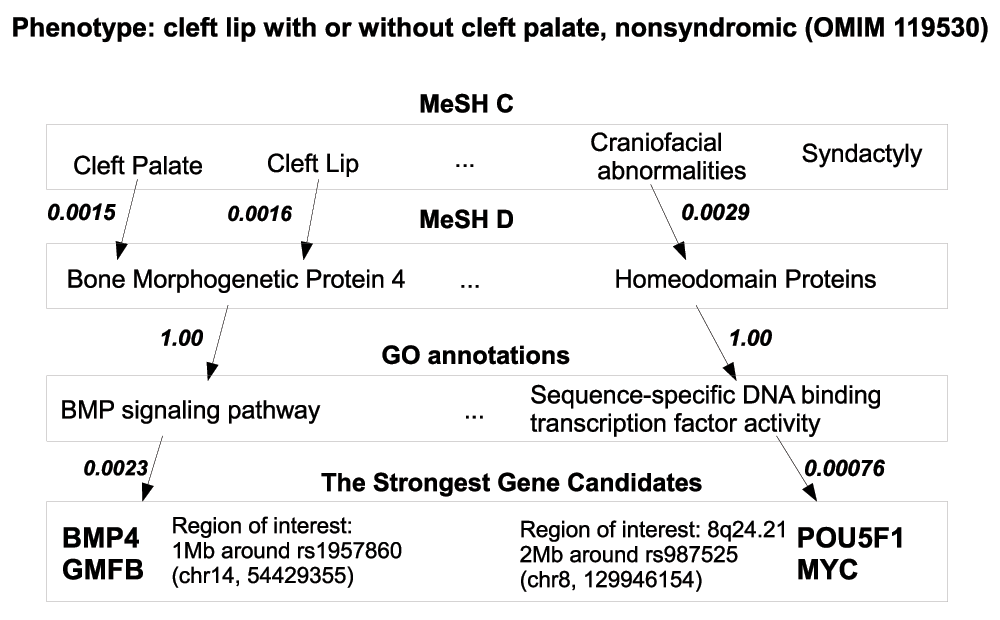
G2D. In G2D the disease phenotype is defined by the OMIM identifier which is mapped to the associated MeSH terms of the diseases. The candidate genes are selected from the provided genomic region possibly containing a marker associated with the disease phenotype. G2D establishes a chain of evidence connecting the disease phenotype to the genes by forming the links between the terms in MeSH and GO annotations. The MeSH terms of the disease (category C) are linked to the MeSH terms of the chemicals and drugs (category D) which are linked to the Gene Ontology annotations. The connections between the terms are established by computing the normalized frequency of PubMed documents in which the MeSH terms (C and D) occur together. The connection between the protein GO annotations and the MeSH D terms are established by computing the normalized frequency of cooccurrence of GO and MeSH D term in papers supporting experimental evidence for the GO annotation. The GO annotation is weighted by a combined score which is used for ranking of the candidate genes.
This inference is illustrated in Figure 1 through the example of exploring candidate genes associated with cleft lip phenotype. Association between the cleft lip and the rs987525 variant from region 8q24.21 has been replicated independently in several different populations55 but no associated gene was found. G2D suggests the MYC gene as candidate. The link between this gene and the cleft lip disease phenotype is inferred through the relationship between the terms “Craniofacial abnormalities” and “Homeodomain proteins” and the relationship between the later term and the GO annotation a “Sequence specific DNA binding transcription factor activity” of the MYC gene. The MYC gene is regulated by the CTCF transcription factor56 which has a binding site at the genomic location of rs987525 leading to a possible hypothesis that this SNP marker might be linked to the cleft lip through a regulatory interaction with the MYC gene57. Another connection between the BMP4 gene and cleft lip OMIM phenotype is inferred through the relationship between the “Cleft lip” term and the term “Bone Morphogenetic Protein 4” which is related to the GO annotation “BMP signalling pathway”. The BMP4 gene harbors the rs1957860 marker variant which is known to be associated with cleft lip58.
Gene prioritization algorithms produce lists of the best candidates which are most strongly related to the phenotype of interest according to the criteria set by the algorithm. Rankings are based on evidence scores of relationships computed by the prioritization algorithm for each candidate gene. For generation of meaningful hypothesis it is important to know what factors led to the obtained rankings and links established between genes and phenotypes. In methods relying on phenotype definitions by training genes a detailed examination of such evidence is difficult. In multipurpose systems such as PolySearch and PosMed provided evidence lacks specificity. Most comprehensive in this respect is G2D in which OMIM phenotype is translated into adequate MeSH term of the disease. In this study an attempt is made to develop means to support computations linking genes and phenotypes defined by the MeSH terms extending beyond the diseases and building upon the algorithmic principles of G2D43,59.
It was shown in applications of Arrowsmith algorithm that biomedical knowledge can be discovered through finding hidden links between concepts in scientific literature. The concepts, co-occurring at high frequency in two disparate sets of literature articles, indicated meaningful links60,61. The link suggested that fish oil can reduce Raynaud’s syndrome symptoms, later confirmed experimentally62. An inference leading to this result was “fish oil reduces blood viscosity, platelet aggregations and vascular re-activity which are increased in Raynaud’s syndrome”63. In similar way algorithms, based on linking the concepts or entities in the collections of data, relate genes to phenotypes by using concept co-occurrences in literature and controlled vocabularies43,64.
Links between phenotypes and gene GO annotations can be computed through intermediate links with chemicals as shown in G2D59. It is hypothesized that phenotype defined by the MeSH term can be meaningfully related to a subset of MeSH D terms denoting molecular entities of drugs and chemicals. Similarly, gene functions encoded by GO annotations can be meaningfully related to molecular entities denoted by MeSH D terms through related chemical processes affecting gene functions. Strengths of relationships can be derived from information in annotations of PubMed articles and NCBI datasets gene2go and gene2pubmed65. Figure 2 outlines the idea of the algorithm in which a phenotype and GO annotations are linked through chemicals.
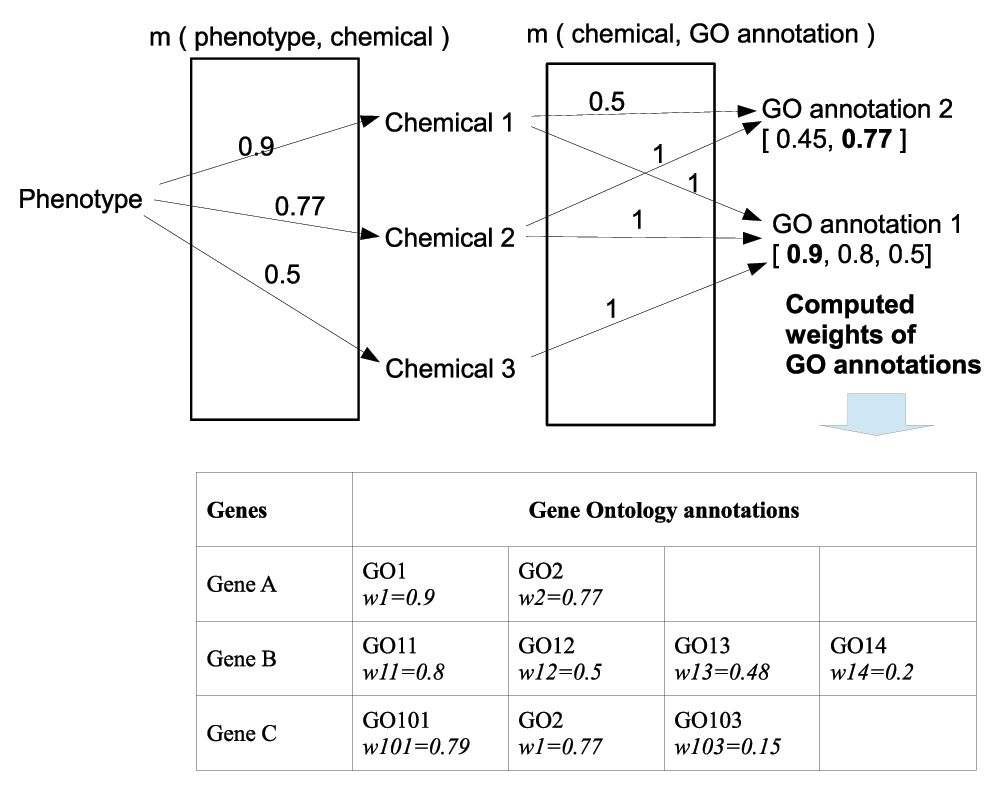
Let us denote MeSH D terms pertaining to chemicals by dj, j = 1, …, N. A relationship m(phenotype, chemical) between the phenotype defined by MeSH term g and the chemical defined by MeSH D term dj is denoted by m(g, dj). Let us denote a relationship m(chemical, GO annotation) between the MeSH D term dj of chemical and GO annotation goi, i = 1, …, M by m(dj, goi). Values of the m(g, dj) and m(dj, goi) relationships represent strengths of the connections between terms. The strengths of connections between the phenotype g and GO annotation goi passing through the chemicals dj, j = 1, …, N are computed as wgoi = maxj (m(g, dj) × m(dj, goi)). These computed weights express the strength of association between the functional annotation goi and the phenotype of interest. Table in a bottom panel of Figure 2 illustrates one possible way to order annotated genes by the magnitude of weights of their association to the phenotype of interest. Principles underlying the algorithm to compute strengths of relationships m(phenotype, chemical) and m(chemical, GO annotation) between phenotypes and functional gene annotations can be founded on fuzzy set theory (FST)43,66. Using mathematical framework of FST the relationships are defined as fuzzy binary relationships (FBRs) and can take a variety of forms67. A thorough explanation can be found in68 on pages 69–84.
Let us denote phenotype MeSH terms as gj, j ∈ (1 … NG) in which j refers to a particular MeSH term. Similarly, let us denote MeSH D terms by dk, k ∈ (1 … ND). A subset of PubMed articles annotated by a specific gj term is denoted by Gj. Similarly, a subset articles annotated by a particular term dk is denoted by Dk. A fuzzy binary relation RGD between two MeSH terms (gj, dk) is defined as:
{[(gj, dk), mgd (gj, dk)] | (gj, dk) ∈ GNG × DND}, (1)
with membership function
The brackets | · | in Equation 2 denote the cardinality, a number of elements in a set, of an intersection |Gj ∩ Dk| of the two sets. The intersection represents a set of the articles annotated by both the gj term and the dk MeSH D term. FBR in Equation 1 is defined on all pairs of selected annotations in the universe of all articles annotated by those MeSH terms. The membership function in Equation 2 models a degree of inclusion of a narrower concept dk (chemical) into a broader concept gj (phenotype) dk ⊆ gj. The FBR of inclusion in a quantitative way defines a semantic relationship between meanings of the broader and narrower concepts69,70.
Inclusion relationship between GO annotations and MeSH D terms is defined using a universe of genes instead of articles. Let us denote GO annotations by goi, i ∈ (1 … NGO) in which i refers to a particular annotation. NGO is a total number of GO annotations of genes in gene2go. Let us denote by GOi a subset of genes in gene2go annotated by a particular goi. Let us denote by GDk a subset of genes in gene2pubmed associated with articles, annotated by the MeSH D term dk ∈ (1 … NGD), where NGD is total number of MeSH D terms associated with genes through articles. Fuzzy binary relation RDGO between these terms is defined as:
{[(dk, goi), mdgo (dk, goi)] | (goi, dk) ∈ GONGO × GDNGD}, (3)
with membership function
The degree of connection between GO annotation and MeSH D chemical in Equation 4 is determined by a number of genes sharing these two annotations over a number of genes annotated by that GO.
A relationship between GO annotation and phenotype defining MeSH term is computed by applying maximum composition operation RGD ○ RDGO on fuzzy binary relations defined by Equation 1 and Equation 3 resulting in a following FBR:
{[(gj, goi), max(mgd(gj, dk) * mdgo (dk, goi))]
| goi ∈ GONGO, dk ∈ (DND ∩ GDNGD), gj ∈ GNG}. (5)
MySQL database and SQL procedures were created in order to experiment with and to support outlined inference70. The created datasets are limited to the annotated genes of human, mouse and fly organisms. The MeSH terms (mtree 2012) defining phenotypes are provided for the categories of Anatomy (A), Diseases (C), Drugs and chemicals (D) and Biological processes and phenomena (G). Information in the created datasets is as of September 2013.
In this section three contributed data sets are described. These datasets and presented MySQL procedure support computations of links between phenotype and GO annotations outlined in a previous section. Datasets were created by using NCBI E-utilities71 and custom scripts. The data sources (as of September 2013) used to create these datasets are described in Table 2. MeSH terms of category B are present but are not used to define phenotype in computation. The datasets are submitted in a format of tab delimited tables that can be imported into MySQL database or used as plain data. In this work a presented data management framework is based on MySQL.
The mesh_terms table stores associations between MeSH terms defining phenotype and MeSH D terms defining chemicals. Statements to create this table in MySQL database are presented in Table 3. Each row stores a pair of MeSH term (category A,C,D and G used to define phenotypes) and a MeSH D term defining a chemical and their relationship as defined by Equation 1 and Equation 2 with supporting information. Data in this table are based on annotated PubMed content as of September 2013. Meaning of columns in mesh_terms table is as follows:
mid is unique identifier of a row;
mterm is MeSH term in which spaces are replaced by underscores (for example Cell_Fusion);
dterm is MeSH D term in which spaces are replaced by underscores (for example BMP4_Protein);
dscore is a float number representing a strength of connection between mterm and dterm computed as in Equation 2;
nm number of PubMed articles annotated by mterm;
nd number of PubMed articles annotated by dterm corresponding to |Ddterm| in Equation 2;
inters number of PubMed articles annotated by both mterm and dterm corresponding to |Gmterm ∩ Ddterm| in Equation 2;
unio number of PubMed articles annotated by either mterm or dterm or both;
pmids comma separated list of PMID identifiers of PubMed articles that are the inters articles;
dtid numerical key identifying dterm in another table dterm_go.
The dterm_go table stores associations between MeSH D terms defining chemicals and gene ontology annotations of genes. This table was created by custom scripts from NCBI gene2go and gene2pubmed datasets. These datasets can be found on NCBI ftp site ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/.
The gene2go dataset stores pairs of gene and its GO annotation. Annotations and genes of human, mouse and fly were retrieved. The gene2pubmed dataset stores pairs of genes and PMIDs of articles associated to them. From this datset pairs of genes and MeSH D annotations of their associated articles were retrieved. These intermediate datasets were used to create the dterm_go table. Statements to create this table in MySQL are presented in Table 4. Meaning of the columns in the dterm_go table is as follows:
dterm is MeSH D term in which spaces are replaced by underscores (for example BMP4_Protein);
goterm is identifier of GO annotation in the Gene Ontology (for example GO:0000001 is identifier for annotation “mitochondrion inheritance”);
gscore is a float number representing a strength of connection between goterm and dterm computed according to Equation 3 and Equation 4;
gogenes number of genes (of mouse, human and fly) annotated by goterm as was recorded in gene2go dataset in NCBI ftp repository corresponding to |GOgoterm| in Equation 4;
genenum number of genes (of mouse, human and fly) sharing the goterm and dterm annotations corresponding to| GDdterm ∩ GOgoterm| in Equation 4;
genetot number of genes (of mouse, human and fly) associated with articles annotated by dterm recorder in gene2pubmed dataset in NCBI ftp repository;
genes comma separated list of Entrez Gene identifiers of genes that form genenum genes;
id unique identifier of the row;
dtid numerical key identifying dterm in table mesh_terms.
Table go_terms stores description information of gene ontology annotations. Statements to create this table in MySQL are presented in Table 5. Meaning of the columns in the dterm_go table is as follows:
gokey is a unique row identifier;
goterm is an identifier of GO annotation in the Gene Ontology (for example GO:0000001 is identifier for annotation “mitochondrion inheritance”);
description is a description of GO annotation in the Gene Ontology in which spaces are replaced by underscores (for example mitochondrion_inheritance is description of GO:0000001 identifier);
category is an indicator of a category of GO annotation and can take value of “Process”, “Function” or “Component”.
Figure 2 and Equation 1, Equation 3, Equation 5 outline a possible way of establishing links between phenotypes defined by MeSH terms and GO annotations that pass through chemicals. These links can be computed by MySQL statements given that MySQL database tables were created as shown in Table 3, Table 4, Table 5. The suggested procedure is presented in Table 6. Three input parameters queryterm, dfrac, gofrac can be provided to the procedure mesh_to_go.sql. This MySQL procedure has a computation and an output part. The parameter gueryterm provides a MeSH term that defines phenotype. 16914 MeSH terms from 2012 MeSH edition72 can be queried in current implementation. These terms form pairs with 5908 MeSH D terms of chemicals. Textual fields of all MeSH terms and GO annotations have underscores instead of spaces between words that should be used in formulating queries.
Parameters dfrac and gofrac set thresholds on the corresponding dscore and gscore values. They can be used to filter terms in computation based on strengths of relationships between the phenotype and chemical and between the chemical and GO annotation (disallowing weaker relationships). Value of dfrac can vary in range of [0.0000041, 1] which is a range of dscore values. Value of gofrac can vary in interval of [0.0001, 1].
In computation presented in Table 6, a creation of t2 and t3 tables corresponds to performing a maximum composition operation defined by Equation 5. The table t2 contains all relationships between the phenotype in queryterm and GO annotations passing through all chemicals that have a connection to the queryterm phenotype. Fewer GO annotations with only maximum weight in relationship to the phenotype are selected into table t3. Statements in the output part create plain sectioned text file. Weighted GO annotations are listed in the first section. Second section identified by “list_of_all_links_go_dterms”, lists all connections in the table t2.
For example, a command line query for phenotype “Intellectual Disability” (user X and database Xdb) can be executed in a following way:
$mysql -u X -p Xdb -e “call \\
mesh_to_go( ‘Intellectual_Disability’,\\
0.01,0.1);” > id_out;The first output file id_out section will have rows:
ms goterm description
0.071428596 GO:0014805 \\
smooth_muscle_adaptation
0.071428596 GO:0045362 \\
positive_regulation_of_ \\
interleukin-1_biosynthetic_process
...
0.060150021 GO:0004908 \\
interleukin-1_receptor_activity
...These GO annotations are weighted by strength of their relationship to the “Intellectual Disability” phenotype. These weighted GO annotations can be used to rank genes as in Figure 2. Second section of output details relationships between the phenotype and chemicals and between the chemicals and GO annotations, for example considering information on GO:0045362:
ms 0.071428596
goterm GO:0045362
description positive_regulation_of_ \\
interleukin-1_biosynthetic_process
gscore 1.0000
dterm Interleukin-1_Receptor_ \\
Accessory_Protein
dscore 0.0714286A connection between “Intellectual Disability” phenotype and MeSH D term “Interleukin-1 Receptor” is quantified by dscore which equals to 0.0714286. Strength of connection between this chemical and “positive regulation of interleukin-1 biosynthetic process” GO term equals to 1. These two values determine the weight ms of this GO term in connection to “Intellectual Disability” (ID) phenotype. This computational procedure with respect to ID phenotype was previously explored73.
In cancer genes accumulate a large number of mutations74 and next generation sequencing screening may produce a vast number of genetic variants and genes. If a gene harboring a variant was not previously reported, then outlined computation can be used to explore connections of that gene to specific cancer based on a current available knowledge.
Among highly mutated genes identified by the whole genome and exome sequencing of breast tumors are PIK3CA, TP53, GATA3, CDH1, RB1, MLL3, MAP3K1 and CDKN1, which were previously observed in clinical breast cancer tumors75. Genes not previously observed in those tumors were TBX3, RUNX1, LDLRAP1, STNM2, MYH9, AGTR2, STMN2, SF3B1. Both sets of genes were explored in relation to “Breast Neoplasms” by ranking genes in whole human genome. The genes were ranked by magnitudes of weights of their annotations with respect to relationship to “Breast Neoplasms” as depicted in Figure 2 employing the outlined computational principle. Top genes from those sets appearing within the Top 5% of the ranked human genome and closer to this interval are listed in Table 7.
Cancer genes are well characterized and widely studied in literature. The genes, not previously reported as carrying clinically important mutations in studies of breast cancer in75 had stronger links with cancer phenotype in question through their GO annotations. The genes from that study: RB1, GATA3, TP53 and CDH1 appeared in high ranking positions. Current exploration identifies “Tamoxifen” being strongly related to the breast cancer phenotype. Such link is logical because this chemical is used to treat hormone-sensitive tumors. Applied computational procedure through relationships between phenotypes and chemicals helps to explore contexts in which biological processes of interest take place. Unexpected links may be discovered that may help to formulate novel biological hypothesis.
As of now, biologists still find it challenging to interpret large lists of poorly characterized genes with algorithms, which are somewhat limited in terms of how they define phenotypes. These lists may originate from a variety of sources, including microarray experiments, ChIP-Seq experiments identifying transcription factors’ target genes, and scientific literature. The algorithm described here is useful in formulating biological hypotheses in situations in which little is known about the phenotype and the genes in question. The algorithm begins by linking lists of gene GO annotations to phenotypes (non-disease and disease) described by meaningful keywords through the MeSH and PubMed databases. The algorithm then deduces which of the links between the genes and phenotypes are strongest and presents the results in an organized manner. This is different from most of existing algorithms in terms of the methods used to define phenotypes of interest and infer their relationships with genes. To better understand how the outlined algorithm is unique, the existing algorithms are parsed into three categories overlapping at some extent and examined.
The first category of existing algorithms only uses known phenotype-related genes, or training genes, while the second focuses solely on human disease phenotypes. The third category uses general keywords from literature to define phenotypes. All must deduce how genes and phenotypes are related by mining selected information sources, retrieving and integrating data from them, but there are some differences between them. Each will be discussed in turn.
Algorithms based on the use of training genes known to be related to the phenotype of interest, as in the Endeavour39 and ToppGene45 tools, make prioritizations on the basis of pattern classification76. Training genes extract phenotype-defining information from various data sources based on how similar the phenotype is to the training genes, and then build a model of a phenotype based on this extraction42. In other words, the model represents gene features that are most characteristic of that specific phenotype. The candidate genes are ranked by how similar their features are to the features of the model. For example, Endeavour relies on genomic data fusion from multiple information sources41. This tool may be very useful if the properties of the training genes clearly define the phenotype properties of interest in the organisms being investigated. Knowing these properties, one can characterize candidate genes by comparing them to the training genes. Genes that have similar characteristics to the training genes may also play an important role in previously unknown phenotype expressions. This principle of discovery is known as “guilt by association”77. Although very useful in detecting similarities between candidates and training genes, the integration of data from multiple sources has limitations. The main limitation is in existing schemes of combining the information from different sources to rank the candidates41. First, the prioritization algorithms using training genes generally differ with respect to the data sources they use23. Different information sources of training genes lead to different models and similarity metrics. Second, some data sources do not have complete information on some genes, so if the phenotype in question has not been sufficiently studied and there are no genes known to be associated with it, then the training genes approach is not effective. Third, the training genes might represent a heterogeneous group biasing phenotype definition in some way. For example, the data fusion scheme relies on the independence of information sources about gene properties, but in practice they are not entirely independent. Protein-protein interaction databases, the gene interaction databases and gene ontology refer to scientific publications as supporting evidence for the information they store, and might even be derived from the literature. While it should do so, the scheme does not always account for these possible interactions and overlaps between sources.
Many tools specific to human diseases use phenotype definitions from databases47,59. Because human disease phenotypes have been extensively studied and are well represented by OMIM78, and because they contain structured information suited to uncovering meaningful links between human diseases and genes, it is relatively easy to associate genes with said phenotypes. However, phenotypes other than diseases and phenotypes in Mammalian Phenotype Ontology37 are not yet represented by well-structured and information-rich resources33,34.
The third category of algorithms, which use general keywords from literature to define phenotypes, are exemplified by tools such as PosMed, PolySearch, GeneProspector and CANDID48,51–53. These tools rely on finding matching documents in MEDLINE or locally-created databases, and then associating genes with the matching documents. General purpose discovery-oriented systems such as iHOP79, Anni2.080, Arrowsmith60,61 and PosMed52, use conceptual networks. Users can browse through the network and create textual profiles describing genes, proteins, or other biomedical concepts. However, once again, there will be genes and processes that are not well represented in literature and there is little information about them that can be retrieved.
Thus, while the obvious advantage offered by specialized gene information databases is that specific information can be extracted very quickly, complementing the literature with more information sources for gene prioritization is advantageous in allowing potential use of algorithms that offer novel interpretations of existing information. G2D43,59 is the only existing method which provides underlying ideas for the algorithmic approach outlined here, which prioritizes genes with respect to human disease phenotypes. However, the scope of the application of the developed algorithm to link genes with phenotypes70 outlined here is distinct from G2D by contributing the following:
The outlined algorithm establishes meaningful links between genes and phenotypes, and enables prioritization, beyond human disease phenotypes by using concepts of MeSH vocabulary from the categories A, D, and G.
The proposed algorithm can be applied beyond human organisms as the annotated genes of the entire genome for human (Homo sapiens), mouse (Mus musculus) and fly (Drosphila melanogaster) are used. In contrast, G2D focuses on human genes.
The data in the gene2go and gene2pubmed NCBI databases65 are used to link GO annotations to MeSH terms of Drugs and Chemicals describing the molecular entities. In contrast, G2D works with the RefSeq database for this purpose43.
The outlined algorithm similarly to G2D utilizes fuzzy binary relationships between concepts, based on mathematical operations of fuzzy set theory67,68, to infer gene-phenotype links. G2D uses a similarity relationship43 while this algorithm uses an inclusion relationship70.
The outlined algorithm is an attempt to remedy some of the challenges presented by information shortages and the way existing algorithms described above are configured to define phenotypes and determine relationships. It has important advantages compared to the other gene prioritization algorithms, in addition to G2D, reviewed extensively in Introduction section.
The approach to link genes and phenotypes outlined in this work represents one out of existing possible approaches. Contributed datasets opens possibility to experimentation and development of other applications. These datasets, although in need of updating comprise co-occurrences of selected categories of MeSH terms in PubMed and co-occurrences of MeSH D terms with GO annotations created from NCBI Gene datasets. Availability of such offline data saves time of a researcher who may want to explore and apply text and data mining algorithms to analyze relationships between concepts.
Existing tools provide limited explanations for reasons for phenotype gene association. Using the outlined approach, evidence supporting the obtained strongest links can be easily examined. As a result of inference, the MeSH D terms which are most strongly related to both the candidate genes through their GO annotations and phenotype are identified. This is useful as it reveals the physical background domains related to the candidate genes gleaned from associated articles without reading their full text. The availability of this background information opens up the possibility of identifying and examining unique aspects of the functions of the studied genes.
However, a single information source cannot account for all aspects of gene relations to phenotypes even if MeSH vocabulary contains information about the processes, phenomena and phenotypes studied in literature. And while functional gene annotations are also associated with scientific publications, there will be genes and processes that are not well represented in literature, as stated earlier. In this situation inferring links between genes and phenotypes might be more effective using other information sources, as genes can also be characterized by their interactions with other molecular entities, by their sequences and by the information about the protein domains of the products. These gene properties can be retrieved computationally from other specialized databases.
F1000Research: Dataset 1. Table mesh_terms, 10.5256/f1000research.6140.d4316782
F1000Research: Dataset 2. Table dterm_go, 10.5256/f1000research.6140.d4316883
F1000Research: Dataset 3. Table go_terms, 10.5256/f1000research.6140.d4317684
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)