Keywords
Perl, next generation sequencing, RNA-seq, read mapping, pipelines
Perl, next generation sequencing, RNA-seq, read mapping, pipelines
We have addressed the reviewers' suggestions and updated the manuscript accordingly at different places. Specifically, we have worked out what is unique about the ViennaNGS suite in the Introduction and added a new Applications section where we put emphasis on the process of building custom NGS analysis pipelines by means of the ViennaNGS tutorials. Moreover, we have benchmarked CPU and memory consumption of our package, providing results in Table 1 and added a paragraph on the internal testing strategy. In addition, we highlight past and future use cases and development plans of the software and mention possible parallelization scenarios in the Discussion.
To read any peer review reports and author responses for this article, follow the "read" links in the Open Peer Review table.
Next-generation sequencing (NGS) technologies have influenced both our understanding of genomic landscapes as well as our attitude towards handling big biological data. Emerging functional genomics methods based on high-throughput sequencing allow investigation of highly specialized and complex scientific questions, which continuously poses challenges in the design of analysis strategies. Moreover, the demand for efficient data analysis methods has dramatically increased. While a typical NGS analysis workflow is built on a cascade of routine tasks, individual steps are often specific for a certain assay, e.g. depend on a particular sequencing protocol.
Here, we present ViennaNGS, a Perl distribution that integrates high-level routines and wrapper functions for common NGS processing tasks. ViennaNGS provides tools and functionality for the development of custom NGS pipelines, rather than being an established pipeline per se. It comes with a set of utility scripts that serve as reference implementation for most library functions and can readily be applied for specific tasks or integrated as-is into tailor-made pipelines. Moreover, we provide extensive documentation, including a dedicated tutorial that showcases core features of the software and discusses common application scenarios.
A set of NGS analysis pipelines are available for general1,2, and specialized assays such as de-novo motif discovery3. While these tools mostly cover the elementary steps of an analysis workflow, they often represent custom-tailored solutions that lack flexibility. Web-based approaches like Galaxy4 cover a wide portfolio of available applications, however they do not offer enough room for power users who are used to the benefits of the command line.
The recently published HTSeq framework5 as well as the biotoolbox package provide library modules for processing high-throughput data. While both packages implement NGS analysis functionality in a coherent manner, we encountered use cases that were not covered by these tools.
ViennaNGS is a pure Perl-based alternative to existing approaches, addressing the broad Perl community in bioinformatics. It partly builds on BioPerl6 and has been designed in an object-oriented manner based on the Moose object framework, thus allowing to write modular code with different libraries that engage with one another. Moose is based in large part on the Perl 6 object system, thererby enabling rapid conversion to Perl 6. While there is ongoing discussion in the BioPerl community regarding possible directions towards a shift to Perl 6, ViennaNGS is, to our knowledge, the first toolbox for NGS data processing that can be regarded ready for Perl 6.
The motivation for this contribution emerged in the course of the research consortium “RNA regulation of the transcriptome” (Austrian Science Fund project F43), which brings together more than a dozen experimental groups with various thematic backgrounds. In the line of this project it turned out that complex tasks in NGS analysis could easily be automated, whereas linking individual steps was very labour-intensive. As such, it became apparent that there is a strong need for modular and reusable software components that can efficiently be assembled into different full-fledged NGS analysis pipelines. Development of the ViennaNGS suite was triggered by two driving forces. On the one hand we wanted to return to the open source community our own contribution, which itself is heavily based and dependent on open source software. On the other hand, beside “open science” we advocate for the concept of “reproducible science”7. Unfortunately, and to some extent surprising, bioinformatics analyses are often not fully reproducible due to inaccessibility of tools (keyword “in-house script”) or software versions used. It is therefore essential to ensure the entire chain of reproducibility from data generation to interpretation in the analysis of biological data.
ViennaNGS has been designed to facilitate the process of builing NGS pipelines. To this end, the toolbox comes with several modules and library functions that can easily be combined into custom analysis workflows. We provide step by guides in the form of dedicated tutorials to lead prospective users through the development of basic NGS analysis pipelines.
ViennaNGS::Tutorial is a showcase for building custom analysis pipelines and consists of several chapters, each illustrating an example workflow together with a possible solution based on ViennaNGS library functions. Tutorial #0 shows how to deduce both qualitative and quantitative parameters from mapped reads, together with visual data representation. Tutorial #1 exemplifies the detection of sequence motifs in close proximity to gene start loci in order to identify regulatory regions. Tutorial #2 exemplifies the visualization of highly expressed genes together with a 50 nt region upstream of the gene start and Tutorial #3 explains how to construct UCSC genome browser Assembly Hubs. The tutorials are meant to assist prospective users applying ViennaNGS to implement their own full-fledged pipelines. Moreover, we used the tutorials to demonstrate the run time and memory requirement of sample implementations of ViennaNGS in a real world scenario (Table 1).
Data were collected on a single core of a desktop workstation (Intel® Core™ i7-4771 CPU @ 3.50GHz; 32GB RAM).
The ViennaNGS suite comes with a collection of complementary executable Perl scripts for accomplishing routine tasks often required in NGS data processing. These command line utilities serve as reference implementations of the routines implemented in the library and can readily be used for atomic tasks in NGS data processing. Table 2 lists the utilities and gives a short description of their functionality.
While some of these scripts are re-implementations of functionality available elsewhere, they have been developed primarily as reference implementation of the library routines to help prospective ViennaNGS users getting started quickly with the development of custom pipelines.
The major design consideration for the ViennaNGS toolbox was to make available modular and reuseable code for NGS processing in a popular scripting language. We therefore implemented thematically related functionality in different Perl modules under the Bio namespace (Figure 1).
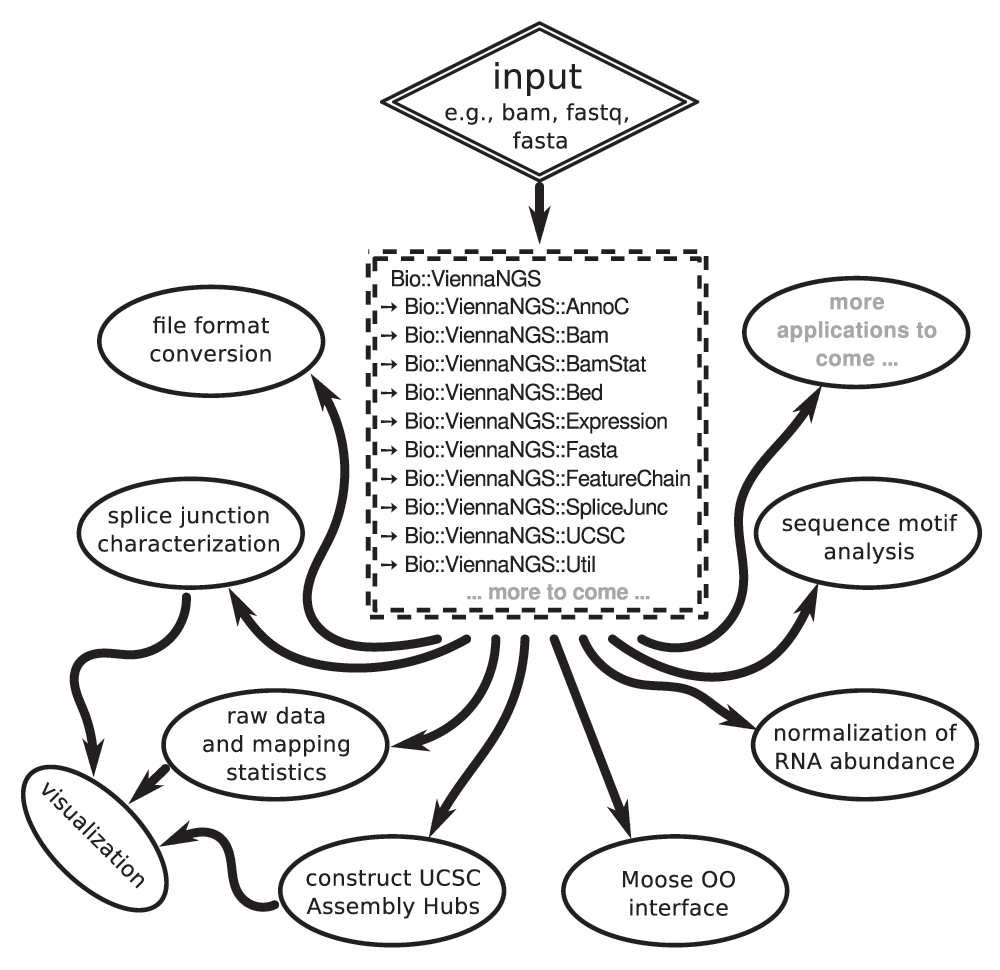
Core modules can be combined within a data analysis script in a flexible manner to meet individual analysis objectives and experimental setup.
Our focus is on consistent versioning, facilitated through Github hosting. In addition, ViennaNGS releases are available via the Comprehensive Perl Architecture Network (CPAN), thereby enabling users to get back to previous versions at any time in order to reenact conclusions drawn from shared biological data.
ViennaNGS has been designed to close gaps in established analysis workflows by covering a wide range of processing steps from raw data to data visualization. In the following we introduce individual ViennaNGS components and describe their main functionality.
Once mapped to a reference genome, NGS data is typically stored in the widely used SAM/BAM file format. BAM is a binary format, which can easily be converted into text-based SAM format via samtools8 for downstream analysis. However, modern NGS assays produce hundreds of millions of reads per sample, hence SAM files tend to become excessively large and can have a size of several hundred gigabytes. Given that storage resources are always limited, strategies to efficiently retrieve mapping information from BAM format are an asset. To accomodate that, we provide functionality for querying global mapping statistics and extracting specific alignment information from BAM files directly.
ViennaNGS::BamStat extracts both qualitative and quantitative information from BAM files, i.e. the amount of total alignments, aligned reads, as well as uniquely and multi mapped reads. Numbers are reported individually for single-end reads, paired-end fragments and pairs missing a mate. Quality-wise ViennaNGS::BamStat collects data on edit distance in the alignments, fraction of clipped bases, fraction of matched bases, and quality scores for entire alignments. Subsequently, ViennaNGS::BamStatSummary compares different samples in BAM format and illustrates results graphically. Summary information is made available in CSV format to facilitate downstream processing.
Efficient filtering of BAM files is among the most common tasks in NGS analysis pipelines. Building on the Bio-SamTools distribution, ViennaNGS::Bam provides a set of convenience routines for rapid handling of BAM files, including filters for unique and multiple alignments as well as functionality for splitting BAM files by strand, thereby creating two strand-specific BAM files. Results can optionally be converted to BedGraph or BigWig formats for visualization purposes.
Proper handling of genomic intervals is essential for NGS analysis pipelines. Several feature annotation formats have gained acceptance in the scientific community, including BED, GTF, GFF, etc., each having its particular benefits and drawbacks. While annotation for a certain organism is often only available in a specific format, interconversion among these formats can be regarded a routine task, and a pipeline should be capable of processing as many formats as possible.
We address this issue at different levels. On the one hand, we provide ViennaNGS::AnnoC, a lightweight annotation converter for non-spliced genomic intervals, which can be regarded a simple yet powerful solution for conversion of bacterial annotation data. On the other hand we have developed an abstract representation of genomic features via generic Moose-based classes, which provide functionality for efficient manipulation of BED4, BED6, BED12 and GTF/GFF elements, respectively, and allow for BED format conversion facilitated by ViennaNGS::Bed. ViennaNGS::MinimalFeature represents an elementary genomic interval, characterized by chromosome, start, end and strand. ViennaNGS::Feature extends ViennaNGS::MinimalFeature by two attributes, name and score, thereby creating a representation of a single BED6 element. ViennaNGS::FeatureChain pools a set of ViennaNGS::Feature objects via an array reference. All intervals of interest can be covered by a ViennaNGS::FeatureLine object, which holds a hash of references to ViennaNGS::FeatureChain objects (Figure 2).
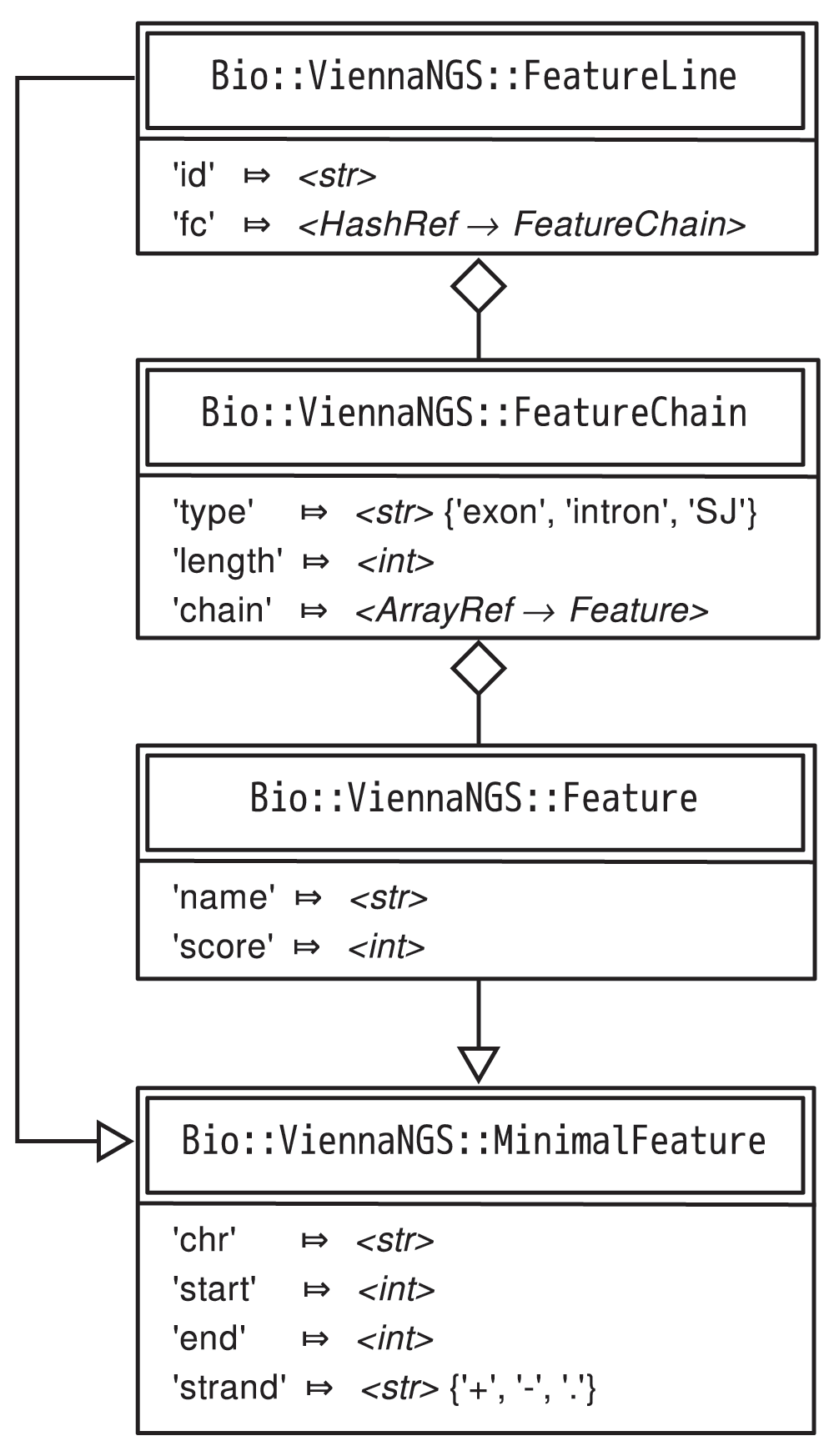
This framework can handle annotation data by providing abstract data representations of genomic intervals such as exons, introns, splice junctions etc. It allows for efficient description and manipulation of genomic features up to the level of transcripts (Figure 3). Conversely, it is highly generic and can be extended to hierarchically higher levels such as genes composed of different transcript isoforms or clusters of paralogous genes.
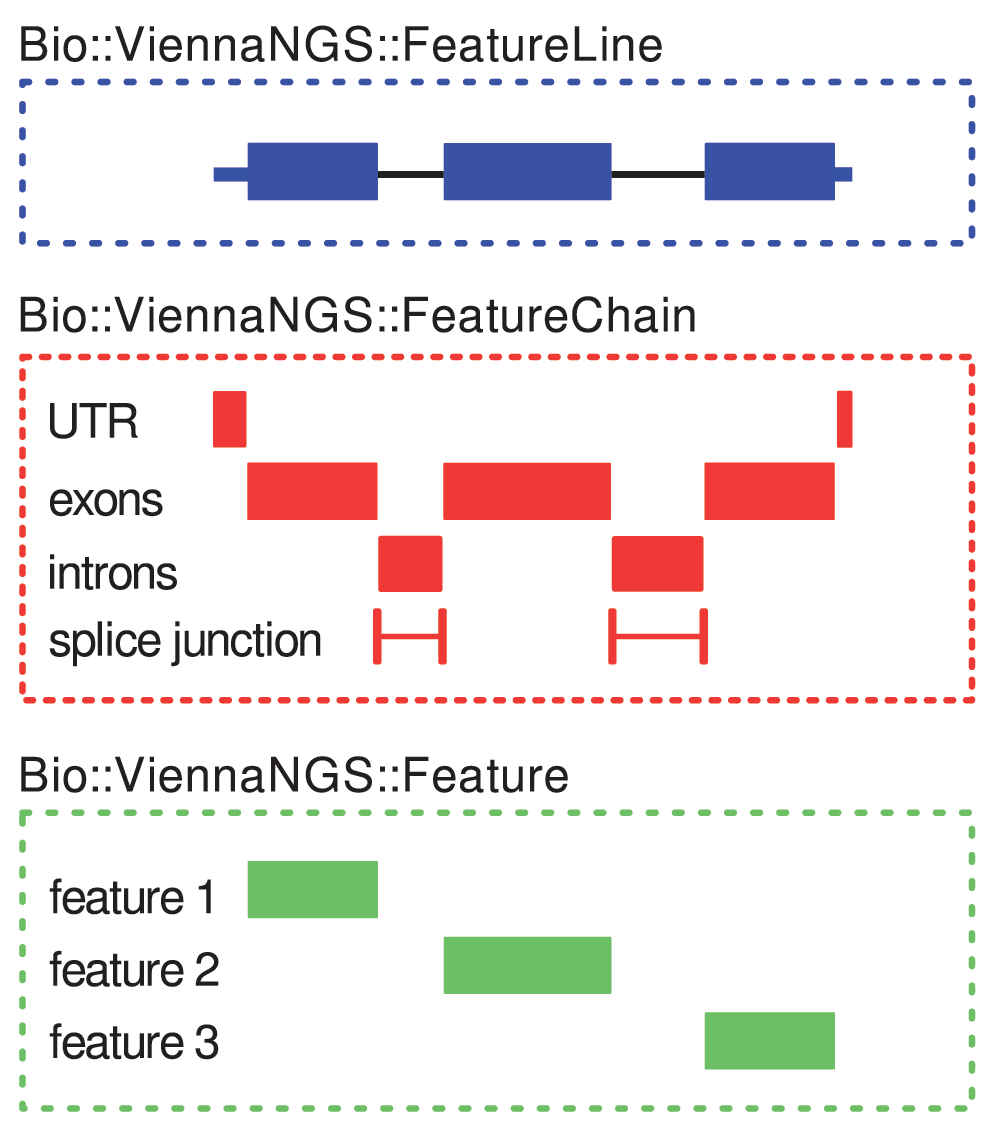
Simple intervals (“features”) are characterized by ViennaNGS::Feature objects (bottom box). At the next level, ViennaNGS::FeatureChain bundles these, thereby maintaining individual annotation chains for e.g. UTRs, exons, introns, splice junctions, etc. (middle box). The topmost level is given by ViennaNGS::FeatureLine objects, representing individual transcripts.
Another cornerstone of NGS analysis pipelines is graphical representation of mapped sequencing data. In this context a standard application is visualization of ChIPseq peaks or RNA-seq coverage profiles. The latter are typically encoded in Wiggle format, or its indexed binary variant, BigWig, which can readily be displayed within a genome browser. In the same line, genomic annotation and intervals are often made available in BigBed format, an indexed binary version of BED. ViennaNGS::Util comes with wrapper routines for automated conversion from common formats like BAM to BigWig or BED to BigBed via third-party utilities9. In addition, we have implemented interfaces for a selection of BEDtools10 components as well as a collection of auxiliary routines. The UCSC genome browser allows to display potentially large genomic data sets, that are hosted at web-accessible locations by means of Track Hubs11. On a more general basis this even works for custom organisms that are not supported by default through the UCSC genome browser, via Assembly Hubs. A typical use case is visualization of genomic annotation, RNA-seq coverage profiles and ChIPseq peaks for Arabidopsis thaliana (which is not available through the generic UCSC browser) via a locally hosted Assembly Hub. ViennaNGS::UCSC provides all relevant routines for automatic construction of Assembly and Track Hubs from genomic sequence and/or annotation. Besides automated Assembly and Track Hub generation, we support deployment of custom organism databases in local mirrors of the UCSC genome browser.
RNA-seq has become a standard approach for gene and transcript quantification by means of measuring the relative amount of RNA present in a certain sample or under a specific condition, thus ideally providing a good estimate for the relative molar concentration of RNA species. Simple “count-based” quantification models assume that the total number of reads mapping to a region can be used as a proxy for RNA abundance12. A good measure for transcript abundance is ideally as closely proportional to the relative molar concentration of a RNA species as possible. Various measures have been proposed, one of the most prominent being RPKM (reads per kilobase per million). It accounts for different transcript lengths and sequencing depth by normalizing by the number of reads in a specific sample, divided by 106. It has, however, been shown that RPKM is not appropriate for measuring the relative molar concentration of a RNA species due to normalization by the total number of reads13,14.
Alternative measures that overcome this shortcoming have been suggested, e.g. TPM (transcript per million), where a proxy for the total number of transcript samples considering the sequencing reads per gene is used for normalization, rather than the total number of mapped reads. We provide routines for the computation of RPKM and TPM values for genomic intervals from raw read counts within ViennaNGS::Expression.
ViennaNGS::SpliceJunc addresses a more specific problem, namely characterization of splice junctions which is becoming increasingly relevant for understanding alternative splicing events. This module provides code for identification and characterization of splice junctions from short read mappers. It can detect novel splice junctions in RNA-seq data and generate visualization files. While we have focused on processing the output of segemehl15,16, the module can easily be extended for other splice-aware split read mappers.
The ViennaNGS suite comes with extensive documentation based on Perl’s POD system, thereby providing a single documentation base which is available through different channels, e.g. on the command line via the perldoc utility or on the Web via CPAN.
In the development process of the ViennaNGS suite special emphasis has been placed on code integrity, thereby ensuring that the software produces correct results as novel features are added and the code base is maintained. To achieve that, we make use of the Perl testing framework, which allows to build automated tests that are run at installation time and highlight any issues with code or third party dependencies. Furthermore this includes comparison of MD5 sums for output files produced by ViennaNGS routines, thereby enabling consistency and reproducibility of biological results.
We have successfully applied components of ViennaNGS in the course of an ongoing, large scale collaboration project focusing on RNA regulation. It has been used with different genomics assays in a wide range of biological systems, including human, plants and bacteria. While we have primarily applied ViennaNGS in combination with the short read aligner segemehl15,16, e.g. in a study addressing ribosome associated mRNA degradation in Drosophila17, it has also been used recently with Tophat18 output in a large scale transcriptome study of Ebola and Marburg virus infection in human and bat cells (Hölzer et al., unpublished data).
ViennaNGS is a comprehensive software library for rapid development of custom NGS analysis pipelines. An aspect that is becoming increasingly relevant in scientific computation is parallelization. While we have focused on code convenience, feature richness and easy extensibility, custom ViennaNGS-based pipelines can potentially be implemented in a parallel manner by the end user, e.g. through the Perl threads functionality. An example would be to process and filter a set of BAM files in parallel, provided sufficient IO resources are available.
ViennaNGS is actively developed and its code base is constantly maintained and expanded. We will provide a generic, Moose based annotation converter that builds on and extends the feature annotation classes in the future. In addition, we will incorporate functionality for manipulation and storage of sequence variants, such as SNPs, editing and modification events. ViennaNGS will also be used for automated UCSC genome browser integration in an upcoming version of TSSAR19, a recently published approach for characterization of transcription start sites from dRNA-seq data. Moreover, we will provide Bio::HubFactory, a ViennaNGS-based Web Service for automatic generation of UCSC genome browser Assembly Hubs for all RefSeq bacteria.
ViennaNGS is an open platform for building specialized NGS pipelines, which fills a niche by providing functionality that is, to our knowledge, not available elsewhere. In this line, we would like to encourage the scientific community to contribute novel features and patches via Github.
Input data for the ViennaNGS tutorial is available from http://rna.tbi.univie.ac.at/ViennaNGS.
The ViennaNGS distribution is available through the Comprehensive Perl Architecture Network (CPAN) and at GitHub.
The ViennaNGS toolbox depends on a set of third-party tools and libraries which are required for specific filtering and file format conversion tasks as well as for building internally used Perl modules:
bedGraphToBigWig, fetchChromSizes and faToTwoBit from the UCSC Genome Browser applications9
samtools <= v0.1.1913 for building Bio::DB::Sam. Please note that more recent HTSlib-based versions of samtools will not work with Bio::DB::Sam
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)