Keywords
Publication bias, research transparency, evidence-based medicine, antidepressants, gabapentin, celecoxib, rosiglitazone, oseltamivir
This article is included in the All trials matter collection.
Publication bias, research transparency, evidence-based medicine, antidepressants, gabapentin, celecoxib, rosiglitazone, oseltamivir
In this revised version, I very slightly changed diction in one area of the antidysrhythmic and celecoxib sections to improve readability. I also provided much additional detail on the oseltamivir meta-analyses for interested readers. Importantly, these revisions merely serve to augment the original version, not to change any messages or statements concerning the evidence.
Recent systematic reviews and meta-analyses from 2009, 2010, and 2013 all confirm the widespread presence of publication bias in the medical literature, with the two most recent studies finding around 50% or more of studies go unpublished.1–3 The failure to publish studies is often due to the studies having so-called “negative” or non-significant findings.1–3 In a similar manner, issues with inadequate research transparency continue to loom large, with important elements of a study changing from the original protocol to the final manuscript that appears in the peer-reviewed literature without proper acknowledgement of such changes in the final manuscript.1,4–6 The existence and persistence of these two closely-related issues carry profoundly detrimental implications for the evidence base from which researchers and medical providers operate. Importantly, these issues put patients at risk of receiving incomplete or inaccurate medical advice and/or an intervention that appears much more effective or advantageous than it really is, is less than ideal for the condition or situation in question, or is actually harmful. Such a claim should not be taken lightly. The systematic reviews and other studies aforementioned1–6 collectively provide important and robust evidence of the incontrovertible existence and pervasiveness of these issues; however, these studies, although incredibly valuable, might still blur somewhat the concreteness of these issues. Therefore, it is instructive, and perhaps even crucial, to go deeper into the “rabbit hole,” where one finds many well-documented specific examples that add unequivocal tangibility to the dire importance and relevance of these issues. To this end, this article presents a “case series” that demonstrates the very real effects publication bias and inadequate research transparency have on the medical literature, and in turn, the very real effects these issues can have on patient care and the practice and furthering of evidence-based medicine.
After suffering a myocardial infarction (MI), some people develop dysrhythmias or aberrant heartbeats. Sometimes this is transient, but in other cases this can portend an increased risk for potentially lethal complications. Indeed, those who do develop persistent dysrhythmias have a higher risk of death than those who do not. Given this common medical knowledge, the historical question was: Should medical providers proactively administer medications (specifically, class I antidysrhythmic agents) that act to suppress dysrhythmias after a patient has suffered a MI (post-MI antidysrhythmic prophylaxis)?
This is precisely what used to be done, but when Furberg completed his 1983 review assessing the impacts of this practice, there was no evidence of benefit from doing this (there was not even a trend suggesting possible benefit); in fact, Furberg even mentioned some emerging evidence that these agents might be harmful in this setting.7 However, he still concluded that the finding of no benefit seemed the least likely possibility, and instead, he concluded the most likely explanation was that the class I antidysrhythmics did offer mortality benefit that was probably obscured by methodological shortcomings of the available trials.7 These agents continued to be used for almost an entire decade after his review. Then, the trials CAST and CAST-II were published in 1991 and 1992, followed by a meta-analysis in 1993.8–10 All these studies found that post-MI antidysrhythmic prophylaxis not only offered no benefit, but also caused substantial harm in the form of excess death.8–10
The harm amounted to the deaths of tens of thousands of people in the 1980s for every year they were used (especially later in the 1980s), with a contextual comparison of the annual number of deaths caused during the peak of their use quite possibly being greater than the number of U.S. military personnel killed in the entirety of the Vietnam War. The upper-level estimate is approximately 65,000 – 70,000 excess deaths per year, and even the most conservative estimate still yields approximately 28,000 excess deaths per year of use. The total excess death estimate is in the hundreds of thousands.11
The story gets worse. In 1993, Cowley and colleagues published an article describing a randomized, double-blind, placebo-controlled trial conducted in 1980, prior to the publication of Furberg’s 1983 review.12 In this study, there were nine deaths in the antidysrhythmic (lorcainide) group of 48 patients (18.75%; the abstract mistakenly indicates there were 49 patients in the lorcainide group, but there were actually 48), and only one death in the placebo group of 47 patients (2.13%).
Although the authors never conducted any statistical analysis on the mortality data, if one were to consider further the mortality event rates (in what is obviously a post hoc analysis), one would find an absolute increase in risk of death of 16.62% over the six-week period of the study for those in the lorcainide group (95% confidence interval [CI], 4.21% – 29.93%), and by Fisher’s exact test, the difference between the two groups would have a P value of 0.015 (rounded to three decimal places). Based on these results, one additional and unnecessary death was caused for about every six patients treated for six weeks with lorcainide for post-MI antidysrhythmic prophylaxis.
Since the development of lorcainide was “abandoned for commercial reasons,” the study was not initially published.12(p166) Though the authors maintain they initially thought the higher incidence of death in the lorcainide group was due to chance, the authors later came to rightly regard this as an “example of publication bias,” and they also recognized that, had the study been properly published, it “might have provided an early warning of trouble ahead.”12(p166)
In 2008, Turner and colleagues published a striking review of 12 antidepressants where they compared all the phase II and phase III trials submitted to the FDA to the corresponding trials available in the peer-reviewed literature.13 The 12 antidepressants included selective serotonin reuptake inhibitors (SSRIs; citalopram, escitalopram, fluoxetine, paroxetine, paroxetine CR, sertraline), serotonin-norepinephrine reuptake inhibitors (SNRIs; venlafaxine, venlafaxine XR, duloxetine), and agents with other mechanisms of action (bupropion SR, mirtazapine, nefazodone). Figure 1 shows the primary results graphically, along with several additional graphical representations of the study’s data. How is this possible? Of the 38 positive studies, all but one were published, yielding 37 positive studies in the peer-reviewed literature. Of the 36 not-positive (24 negative and 12 questionable) studies, three were correctly published as negative, 22 were not published at all, and 11 were published in such a way as to make the results seem positive (a manner of reporting referred to as “spinning”).

All graphs were created using data from Turner and colleagues’ 2008 publication. Percentages are rounded to nearest tenth of a percentage point.13
Interestingly, Melander and colleagues published similarly disconcerting findings in 2003 when they found troubling issues with the SSRI literature compared to the data available at the Swedish drug regulatory authority.14 Out of 42 studies submitted to the drug regulatory authority, 21 showed a significant effect for the primary outcome (SEPO) and 21 found no significant effect for the primary outcome (NSEPO). However, in the peer-reviewed literature, there were 22 corresponding stand-alone publications finding SEPO; interestingly, three of the 21 studies finding SEPO were published as a stand-alone publication twice, with two of the 21 studies finding SEPO never appearing as a stand-alone publication (their data were pooled with other studies for a pooled-results publication). On the other hand, of the 21 studies finding NSEPO, only six were published as a stand-alone publication, the data from four never appeared in any fashion in the peer-reviewed literature, and the remaining 11 only had their data presented in a pooled-results publication incorporating data from other studies. All but one of the pooled-results publications had an amalgamation of studies finding SEPO and NSEPO, with one pooled-results publication being comprised of only two studies finding NSEPO. Additionally, the pooled publications were not always forthright about pooling. For instance, two of the pooled publications for one drug were reported as a “double-blind comparison” and “a large multicentre [sic] study.”14(p2) If this were not already enough, Melander and colleagues also noted that only two of the stand-alone publications presented both an intention-to-treat (ITT) and per-protocol (PP) analysis. The remaining publications presented only one analysis, which tended to be the typically-more-treatment-favorable PP analysis (only 24% of the stand-alone publications reported an ITT analysis). Melander and colleagues rightly conclude:
“[F]or anyone who relies on published data alone to choose a specific drug, our results should be a cause for concern. Without access to all studies (positive as well as negative, published as well as unpublished) and without access to alternative analyses (intention to treat as well as per protocol), any attempt to recommend a specific drug is likely to be based on biased evidence.”14(p4)
Various aspects of the mishandlings of studies for off-label uses of gabapentin are well-documented in NEJM, Trials, and PLoS Medicine.15–17 These studies primarily surrounded treatment of painful conditions (migraine prophylaxis and neuropathic and nociceptive pain), but a smaller portion of the studies investigated use of gabapentin as an adjunctive agent in bipolar disorder. Mishandlings of the data essentially all revolve around not publishing data from studies in a forthright and transparent manner, with infractions such as delaying publication of negative results, never publishing negative results as a full research article, or altering study protocols (particularly study outcomes) without adequate transparency when doing so.15–17 For instance, primary outcomes specified in protocols were sometimes not reported in the corresponding publication or were reported as secondary outcomes; likewise, primary outcomes that did not appear anywhere in the original protocol were added to publications. Proper disclosure of such changes in the primary outcome was variable (even though it should have been universal when it occurred). Additional concerning findings included discrepancies in the results of statistical analyses between internal research reports and journal publications, and there were even discrepancies in the analytic methods used, such as varying definitions of ITT analysis, with one article even noting “In no case did the documents describe an ITT analysis according to the widely accepted definition [of analyzing] all randomized participants in the groups to which they were assigned.”17(p10) Internal company documents provided in the Trials publication and associated additional file are also distressing (Figure 2).16 These documents add an unequivocal degree of tangibility to the issues of publication bias and inadequate research transparency, and it is worth noting these documents and others that contributed to these three publications became available to the public as a result of litigation over fraudulent sales practices in its marketing of Neurontin® (gabapentin).15–17

These and other documents are provided in the open-access Trials article and the associated additional file. Page numbers provided with the images shown here are for the supplemental file.16
The randomized, double-blind CLASS trial purportedly established the gastrointestinal safety advantage of celecoxib (Celebrex®), a cyclooxygenase-2 (COX-2) selective, non-steroidal, anti-inflammatory drug (NSAID), over other nonselective NSAIDs.18 While initially widely heralded, discussed, and disseminated, there are considerable issues with the CLASS trial: the JAMA publication of the CLASS trial reports six-month data for a single trial, but in reality, there were two trials that were pooled together, and the trials ran for 65 weeks and 52 weeks, respectively.18–25 The stark difference is odd and should immediately give one pause and a desire to clarify the differences. Indeed, upon reviewing the FDA literature, one finds there were originally two protocols: N49-98-02-035 (dated January 26, 1998) and N49-98-02-102 (dated August 24, 1998).20,21 Following a total of eight protocol amendments (all of which took place with blinding preserved), the protocol pre-specified the trials were to run for the aforementioned durations of follow-up or until a pre-specified number of events had occurred (20 in each trial or 45 between the two trials).20,21 The protocol also pre-specified a plan to pool the data for the patients receiving celecoxib.20,21 The events accrued at rates lower than predicted, which affected the duration for which the trials ran; indeed, one of the amendments was to lengthen the study period of one of the trials by three months in order to reach the target number of events.21
The story does not end there, though: When all the data were analyzed according to the pre-specified protocol instead of how the authors reported the six-month data in JAMA, the rate of clinically-significant upper gastrointestinal events (CSUGIEs) in celecoxib recipients versus those who received a comparator nonselective NSAID (ibuprofen or diclofenac) was not statistically significantly different.19–25 The authors’ attempt to reply to reader criticism failed to substantiate their presentation of the truncated data, which they should have known would be the case since the FDA reviewers also pointed out why analysis of the data as it is presented in JAMA is inappropriate.19,20,22–25 For example, in the review he prepared for the FDA, Goldkind stated plainly that the post hoc decision to report six-month data instead of all the relevant data was “not statistically valid or supportable,” and he therefore decided “[b]ased on the lack of adequate rationale, these post hoc analyses will not be further discussed or presented in this review.”20(p51) Lu similarly called the decision to report the truncated six-month data “not valid.”19(p8)
Furthermore, the FDA literature shows that the original primary outcome event was defined as CSUGIEs, which was further defined as perforation, upper gastrointestinal bleeding, or gastric outlet obstruction.19–21 However, celecoxib failed to show significant benefit for CSUGIEs even in the methodologically-invalid, post hoc, six-month truncation analysis.18–21 Table 1 below provides the CSUGIE incidence rate data from the FDA literature for the entire study period and for the untenable six-month truncation point. Only by combining CSUGIEs with symptomatic gastroduodenal ulcers that did not cause a CSUGIE (GDUs) did celecoxib demonstrate a statistically significant benefit versus the pooled NSAIDs group, and the addition of GDUs to the original primary outcome of CSUGIEs was another post hoc decision not transparently addressed in the JAMA publication.18–21 Nevertheless, the inappropriately truncated six-month data for this post hoc altered primary outcome are saluted by the authors in their publication.18 Table 2 below provides the incidence rate data from the FDA literature for the post hoc altered primary outcome of CSUGIEs plus GDUs. It is also important to note that GDUs are a clinically weaker and logistically more problematic outcome to use as part of a primary outcome measure compared to the relatively much more robust composite outcome of CSUGIEs. Goldkind gives a succinct and telling overview of the problems inherent in including GDUs as a part of the primary outcome measure on page 22 of his review for the FDA.20
Also of interest is FDA documentation indicating that the sponsor specified original intent to compare the event rate in the group receiving celecoxib (n = 3,987) to each of the groups receiving diclofenac (n = 1,996) and ibuprofen (n = 1,985) separately as long as the outcome data for the pooled celecoxib group were significantly different from the pooled NSAID group (this stepwise approach was employed to prevent type I error).19,20 However, if one reads the JAMA article, one only finds data for the celecoxib group versus the pooled NSAID group, even though the authors noted a significant difference for the celecoxib group versus the pooled NSAID group for the post hoc altered primary outcome.18 This too is peculiar, and it is troubling to note that in the data available in the FDA documents, celecoxib repeatedly failed to separate significantly from diclofenac.19–21 Indeed, celecoxib was not significantly different from diclofenac regardless of whether one assessed CSUGIEs or CSUGIEs plus GDUs, this remained the case for both outcomes when considering the untenable six-month analysis and the analysis over the entire period of data collection, and it still remained the case in additional subgroup analyses (that were not pre-specified) that considered only those who did not take aspirin.19–21 (The aspirin subgroup analysis is rightly questioned by Lu, who noted “at least 34” subgroup analyses were conducted in an exploratory manner “with no pre-specified plan of statistical inference,” and the lack of a priori specification of subgroup analyses based on aspirin is made even more problematic given the failure to show statistical significance in the original primary endpoint.19(p9)) The failure of celecoxib to demonstrate significant benefit compared to diclofenac in the aforementioned analyses should again give one pause. Indeed, as Witter noted in his review for the FDA: “Therefore, particularly for making drug class (i.e. COX-2 vs. NSAIDs) comparisons, it could be argued that beating one NSAID does not mean you beat them all, but losing to one NSAID (or failing to beat it) is losing to them all.”21(p76)
There is still more to the story. As shown below (Figure 3), internal company documents suggest additional troubling behavior.26–28 However, some of the documents also show that some expressed concern that presentation of the truncated six-month data amounted to “cherry picking” (Mona Wahba)26(p8115) or “data massage … for no other reason that [sic] it happens to look better” (Emilio Arbe).26(p8112) Arbe also indicated he “wouldn’t feel too comfortable presenting a fudged version of the facts.”26(p8113)
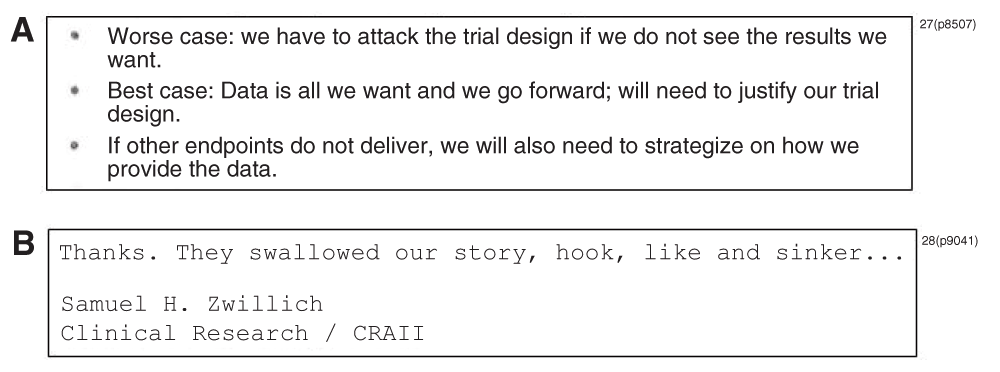
The items above show content regarding strategic planning on what would be done with the celecoxib data based on the favorability of the results and a research director’s response after learning that a symposium offered during a medical conference claimed the CLASS trial demonstrated celecoxib’s superior gastrointestinal safety profile compared to nonselective NSAIDs.27,28
In the trial data discussed above, the dosing regimen of the medications was as follows: celecoxib, 400 mg twice a day; diclofenac, 75 mg twice a day; and ibuprofen, 800 mg three times a day. While the doses of diclofenac and ibuprofen are at the higher end of officially-recommended doses for treatment of pain caused by osteoarthritis and rheumatoid arthritis, the dose of celecoxib used in the trials was two to four times higher than the doses officially recommended for the same indications, respectively. FDA documents indicate the celecoxib dose was deliberately chosen to “ensure that the ulcerogenic potential of celecoxib was rigorously assessed.”21(p10) Unfortunately, instead of providing a rigorous and reassuring assessment, comparing the FDA literature and internal company documents to the official CLASS publication leaves one with little more than profound disillusionment and considerable uncertainty.
Might doses such as 100–200 mg twice daily – the doses typically used in clinical practice – fare differently for similar outcomes? Unfortunately, there is no reliable answer to this question. Some might cite SUCCESS-I, CONDOR, and GI-REASONS (the other major trials of celecoxib versus nonselective NSAIDs) as providing the answers, but doing so is mistaken. For example, the SUCCESS-I trial was a randomized, double-blind trial studying celecoxib at two doses – 100 mg twice daily (n = 4,393) and 200 mg twice daily (n = 4,407) – versus the nonselective NSAIDs naproxen at 500 mg twice daily (n = 905) and diclofenac at 50 mg twice daily (n = 3,489); however, amidst limitations to the study, perhaps most notably for the discussion at hand is the fact that exposure to the medications only lasted 12 weeks, thus precluding any conclusion about long-term effects.29 CONDOR was a randomized, double-blind trial that studied celecoxib 200 mg twice daily (n = 2,238) versus diclofenac slow release 75 mg twice daily plus omeprazole 20 mg once a day (n = 2,246).30 The CONDOR trial ran for six months, which, although better than 12 weeks, is still a disappointing and questionable duration given the aforementioned discussion of the CLASS trial data. This trial has other limitations, but again foregoing such considerations for the discussion at hand, it is still instructive to appraise the actual event rates and the components of the primary composite outcome for the six-month duration of the trial. Notably, the rates of gastroduodenal hemorrhage; gastric outlet obstruction; gastroduodenal, small-bowel, or large-bowel perforation; small-bowel hemorrhage; and large-bowel hemorrhage were identical between the two groups. As shown in Table 3 below, the statistically significant difference reported for the primary composite outcome was driven predominantly by the study-defined outcome of clinically-significant anemia of presumed occult gastrointestinal origin, including possible small-bowel blood loss, with a smaller contribution from clinically-significant anemia judged to be from a visualized gastrointestinal lesion. Similar findings are seen in the GI-REASONS trial, although it had a smaller difference in the primary composite outcome.31 GI-REASONS was another randomized trial that ran for six months, but it was open-label with blinded endpoint assessment and assessed celecoxib (n = 4,035) versus freely-chosen nonselective NSAIDs (n = 4,032). It too has other limitations, but setting those issues aside again to consider the six-month event rates, we again see a statistically significant difference reported for the primary composite outcome (the composition of which was very similar, but not identical, to the primary composite outcome seen in the CONDOR trial). However, as seen in Table 4 below, the difference in the primary composite outcome was again mostly due to differences in clinically-significant anemia of presumed occult gastrointestinal origin, including possible small bowel blood loss, and in this trial, the difference in clinically-significant anemia judged to be from a visualized gastrointestinal lesion was notably smaller, differing by only two events between the groups. Although CONDOR and GI-REASONS had similar durations and outcomes (thereby perhaps tempting some to compare the event rates for the primary composite outcomes to one another), one must note there were differences in methodology (e.g. double-blind in CONDOR versus open-label with blinded outcome assessment in GI-REASONS, fixed-dose and fixed-drug comparison in CONDOR versus clinician-determined dosing and allowance for use of and switching between different nonselective NSAIDs in the nonselective NSAID group in GI-REASONS) and patient population (e.g. exclusion of patients who tested positive for Helicobacter pylori in CONDOR versus allowance of such patients in GI-REASONS). Still, perhaps the most salient and troubling feature is that no trial has ever continued long enough to resolve lingering uncertainties raised by the full dataset behind the CLASS trial regarding the long-term assessment of celecoxib compared to nonselective NSAIDs, and simply meta-analyzing the available trials cannot resolve this uncertainty, either. Finally, all of these trials had significant conflict of interest with respect to trial funding from the parent company for celecoxib and/or financial relationships with the parent company for celecoxib, up to and including working exclusively for the parent company. Although this is not necessarily an incorrigible flaw by default, one is prudent to be cognizant of such conflicts of interest, and given the mishandlings of the CLASS trial, some might reasonably be left with additional uncertainties about the integrity of the conduct of the SUCCESS-I, CONDOR, and GI-REASONS trials.
| Celecoxib (n = 2,238) | Diclofenac slow release plus omeprazole (n = 2,246) | |
|---|---|---|
| Primary composite outcome: clinically-significant events throughout the GI tract* | 20 (0.894%) | 81 (3.606%) |
| Clinically-significant anemia^ of presumed occult GI origin, including possible SB blood loss | 10 (0.447%) | 53 (2.360%) |
| Clinically-significant anemia^ with a defined GI lesion judged to be the cause | 5 (0.223%) | 24 (1.069%) |
| GD ulcer or erosions | 5 (0.223%) | 20 (0.890%) |
| Early gastric cancer | 0 | 1 (0.045%) |
| Lower GI bleeding# | 0 | 1 (0.045%) |
| Lower GI ulcer or erosions | 0 | 2 (0.089%) |
| Acute GI hemorrhage of unknown origin, including presumed SB hemorrhage | 1 (0.045%) | 0 |
| Identical rates for the following components of the primary composite outcome: GD hemorrhage; gastric outlet obstruction; GD, SB, or LB perforation; SB hemorrhage; and LB hemorrhage | ||
Data presented are number (percentage) of patients in each group and come from Table 2 of the CONDOR trial.30
GD indicates gastroduodenal; GI, gastrointestinal; LB, large-bowel; SB, small-bowel.
*The primary composite outcome included: gastroduodenal, small-bowel, or large-bowel hemorrhage; acute gastrointestinal hemorrhage of unknown origin, including presumed small-bowel hemorrhage; gastroduodenal, small-bowel, or large-bowel perforation; gastric outlet obstruction; clinically significant anemia with a defined gastrointestinal lesion judged to be the cause; and clinically-significant anemia of presumed occult gastrointestinal origin, including possible small-bowel blood loss.
^Clinically-significant anemia was defined in the protocol as a decrease in hemoglobin by 2 g/dL or more or a drop in hematocrit of 10% or more.
#One patient in the diclofenac slow release plus omeprazole group was found to have bleeding angiodysplasia in the colon.
| Celecoxib* (n = 4,035) | Nonselective NSAID* (n = 4,032) | |
|---|---|---|
| Primary composite outcome: clinically-significant upper and/or lower GI events^ | 54 (1.338%) | 98 (2.431%) |
| Clinically-significant anemia# of presumed occult GI origin, including possible SB blood loss | 44 (1.090%) | 75 (1.860%) |
| Symptomatic ulcers+ | 0 | 5 (0.124%) |
| LB hemorrhage | 3 (0.074%) | 6 (0.149%) |
| Clinically-significant anemia# with a defined GI lesion judged to be the cause | 4 (0.099%) | 6 (0.149%) |
| Acute GI hemorrhage of unknown origin, including presumed SB hemorrhage | 1 (0.025%) | 3 (0.074%) |
| GD hemorrhage | 0 | 2 (0.050%) |
| Gastric outlet obstruction | 1 (0.025%) | 0 |
| Identical rates for the following components of the primary composite outcome: SB hemorrhage; SB obstruction; GD, SB, or LB perforation | ||
Data presented are number (percentage) of patients in each group and come from Table 1 from the GI-REASONS trial.31
GD indicates gastroduodenal; GI, gastrointestinal; LB, large-bowel; SB, small-bowel.
*The trial allowed dose titration in each group and unrestricted medication selection and switching within the nonselective NSAID group (switching between the groups was not allowed, however). The celecoxib dose most commonly prescribed initially was 200 mg daily (90%). The nonselective NSAIDs included meloxicam (average dose 13.0 mg), naproxen (average dose 819.8 mg), nabumetone (average dose 1,089.0 mg), diclofenac (average dose 124.4 mg), ibuprofen (average dose 1,453.2 mg), and etodolac (average dose 709.2 mg).
^The primary composite outcome included: gastroduodenal, small-bowel, or large-bowel hemorrhage; acute gastrointestinal hemorrhage of unknown origin, including presumed small-bowel hemorrhage; gastroduodenal, small-bowel, or large-bowel perforation; gastric outlet or small-bowel obstruction; symptomatic ulcers; clinically significant anemia with a defined gastrointestinal lesion judged to be the cause; clinically-significant anemia of presumed occult gastrointestinal origin including possible small-bowel blood loss.
#Clinically-significant anemia was defined in the protocol as a decrease in hemoglobin by 2 g/dL or more or a drop in hematocrit of 10% or more.
+Ulcers without complications, presenting with dyspepsia and having endoscopic or standard radiograph evidence of a gastric ulcer and/or duodenal ulcer.
Rosiglitazone (Avandia®) provides us with yet another troubling example.32–43 When John Buse (a physician researcher from the University of North Carolina at Chapel Hill) raised concern over the cardiovascular risk of rosiglitazone starting in 1999, the parent company of the drug took considerable efforts to try to silence him, including complaining to his superiors and threatening legal action. This ultimately ended in the U.S. Senate Committee on Finance (USSCoF) getting involved. The USSCoF eventually determined the actions of the parent company amounted to “intimidation,” and the USSCoF review of the matter is indeed telling:
“The effect of silencing this criticism is, in our opinion, extremely serious. At a July 30, 2007, safety panel on Avandia, FDA scientists presented an analysis estimating that Avandia caused approximately 83,000 excess heart attacks since coming on the market. Had GSK considered Avandia’s increased cardiovascular risk more seriously when the issue was first raised in 1999 by Dr. Buse, instead of trying to smother an independent medical opinion, some of these heart attacks may have been avoided.”32(p2)
Furthermore, additional internal company documents show more duplicitous behavior surrounding rosiglitazone (Figure 4).33,34

H-H indicates head-to-head trial; RSG, rosiglitazone. The images from pages 21 and 22 are from a consecutive email chain.
Once again, the story does not end there. In 2007, Steven Nissen and Kathy Wolski sought to publish a meta-analysis of 42 studies on rosiglitazone in NEJM; however, one day after they submitted it to NEJM, Steve Haffner – one of the pre-publication peer reviewers and also a consultant for GlaxoSmithKline (GSK) – leaked the pre-publication manuscript to GSK in a fax marked “confidential” and “urgent.”35(p5) This unethically leaked manuscript was then disseminated within GSK, and a GSK statistician attempting to find deficiencies in the Nissen and Wolski meta-analysis ultimately found “there is no statistical reason for disregarding the findings as presented.”35(p6)
What exactly did the Nissen and Wolski meta-analysis find? Compared to the control group (defined in their meta-analysis as those who received either placebo or other medications used to treat diabetes), those who received rosiglitazone experienced significantly more MIs, with an odds ratio (OR) of 1.43 (95% CI, 1.03 – 1.98; P = 0.03).36 Nissen and Wolski also assessed death from cardiovascular causes, finding an OR of 1.64 (95% CI, 0.98 – 2.74; P = 0.06).36
GSK and the FDA had also conducted their own meta-analyses, both of which were consistent with the findings in the Nissen and Wolski meta-analysis.35 In a concerned email to company executives on May 8, 2007, Moncef Slaoui, the head of research at GSK, spoke of the meta-analyses:
“-FDA, Nissen and GSK all come to a comparable conclusion regarding increased risk for ischemic events, ranging from 30% to 43%!
-FDA and Nissen (but no final data from GSK date [sic]) reach the conclusion of an HR for death (CHF + IHD) of 1.72 or 1.75!”35(p164)
Despite this, when the Nissen and Wolski meta-analysis was published online on May 21, 2007, GSK responded the same day with the following: “GSK strongly disagrees with the conclusions reached in the NEJM article, which are based on incomplete evidence and methodology that the author admits has significant limitations.”35(p173) This press release is in rather obvious contrast to internal discussions.
Also of note, 27 of the 42 studies included in the Nissen and Wolski meta-analysis were unpublished but available to them as a result of litigation surrounding another drug manufactured by GSK (one of the 27 unpublished studies is also available through the FDA, as it was submitted to the FDA as part of the approval process for rosiglitazone).36 Nissen and Wolski updated their meta-analysis in 2010, which now included a total of 56 trials and importantly included the RECORD trial, an open-label, randomized, controlled trial that is still shrouded in some controversy.35,37–42 The 2010 meta-analysis included 36 unpublished trials (again with one trial also being available through the FDA) and 20 published trials.41 The effect estimate for MI (OR, 1.28; 95% CI, 1.02 – 1.63; P = 0.04) was similar, albeit somewhat less than that seen in the 2007 meta-analysis; with exclusion of the RECORD trial, the effect estimate increased (OR, 1.39; 95% CI, 1.02 – 1.89; P = 0.04) and more closely approximated the 2007 findings.36,37,41
Briefly, interim results from the RECORD trial (which was originally planned to run until 2009) were published in 2007 shortly after Nissen and Wolski’s 2007 meta-analysis.35,37 However, it appears GSK made this decision unilaterally, as opposed to leaving that decision in the hands of the steering committee (SC).35 A GSK employee is also on record reiterating the force with which GSK would pursue the matter with the SC if necessary:
“[I]f the SC believe that publishing interim data will fatally damage their ability to bring the study to a completion- Frank and I will bring that opinion with reasons back to GSK, before pursuing the line- that a decision has been made- live with it”.35(p184)
The SC was convinced, however, and the unplanned interim analysis was published. However, as the pre-publication manuscript was being sent amongst the primary authors, one author noted: “The HR ratio [sic] (and 95% CI) for MI in RECORD is not inconsistent with Nissen’s – and he had more events; what’s to stop him adding the events from RECORD to his meta-analysis and re-enforcing his view?”35(p197) (note this is exactly what happened in the updated meta-analysis published in 2010). This same author also wrote in the same email: “[m]anuscript looks to me to play down 239% INCREASE in HF. I have taken the liberty of doing some rewording.”35(p197) Though one is glad to see the concern about downplaying an increase in heart failure, the tone of the manuscript was still far too strong, as evidenced by the pre-publication editorial and peer review process. Though there is unavoidable uncertainty that accompanies interpretation of an unplanned interim analysis, the published data were not inconsistent with the 2007 Nissen and Wolski meta-analysis.35,37,38 However, the interim analysis was too underpowered to determine anything robustly, a fact that USSCoF documents suggest GSK knew.35,37,38
One peer reviewer plainly noted that the data “are inconclusive about the question of increased risk in the rosiglitazone arm,”35(p201) and an editor for NEJM noted:
“This reviewer, along with other reviewers, asks that you modify the language in multiple locations in the manuscript to tone down your conclusions. This is especially important given that this is an unplanned interim analysis of an ongoing trial, a fact that introduces additional uncertainty. Please note that, in the opinion of all readers, the data that you present are completely compatible with the results of the meta-analysis by Nissen and the meta-analysis for myocardial ischemic events posted on the GSK Web site.”35(p201)
and
“The editors feel strongly that your data do not support the statement that the RECORD results for MI contradict the Nissen meta-analysis; this statement must be removed or modified.”35(p202)
Upon making the requisite changes, the interim RECORD trial results were published in NEJM (electronic publication in June 2007, print publication in July 2007); however, it was accompanied by three editorials, all of which expressed reservations about the publication, including noting it was underpowered, possibly suffered from incomplete event ascertainment, and did not really provide any assurance of the safety of rosiglitazone.37–40
A purportedly independent reevaluation of the RECORD trial cardiovascular outcomes data was also recently published by a team from the Duke Clinical Research Institute (DCRI).42 Although it essentially upheld the interim RECORD analysis, one notes that GSK was allowed to prepare the materials for the reanalysis, and “[d]espite a concerted effort to obtain additional information, only a modest amount of additional person-years of follow-up was obtained (328 person-years)” by the DCRI team, which was mostly “about vital status and not nonfatal MIs or strokes.”42(p247) To give context, there were a total of 25,833 person-years of follow-up for mortality and 23,692 person-years of follow-up for the composite of cardiovascular death, MI, or stroke.42
The aforementioned role that GSK had in the DCRI team’s analysis raises difficult questions. The DCRI team can only operate on the data to which they had access, which was overwhelmingly prepared and provided by GSK. Since Buse first raised concerns in 1999, the above seems to make it clear that GSK’s history with rosiglitazone does not reflect one of scientific integrity or transparency. In its concluding remarks, the 2010 USSCoF document notes:
“The totality of evidence suggests that GSK was aware of the possible cardiac risks associated with Avandia years before such evidence became public. Several years prior to Nissen’s study, it can be argued that GSK was on notice that Avandia may have problems. Based on this knowledge, GSK had a duty to sufficiently warn patients and the FDA of its concerns in a timely manner. Instead, GSK executives intimidated independent physicians, focused on strategies to minimize findings that Avandia may increase cardiovascular risk, and sought ways to downplay findings that the rival drug ACTOS (pioglitazone) might reduce cardiovascular risk. In recent years, pharmaceutical companies have committed acts that forced them to pay the largest criminal fines in American history.”35(p15)
Additionally, even if one were to accept the RECORD trial results, the 2010 meta-analysis by Nissen and Wolski that included the RECORD trial still stands, and there is still the concern over the increased incidence of heart failure in the RECORD data, so it remains clear the RECORD data ultimately do not resolve concerns about rosiglitazone.
In 2012, GSK officially pled guilty to, among other things, failing to report safety data about rosiglitazone to the FDA.43
On July 14, 2009, Japanese pediatrician Keiji Hayashi astutely pointed out a problem with the then-current Cochrane review on oseltamivir (Tamiflu®) in healthy adults.44 Specifically, Hayashi noted the Cochrane review based its conclusion about the effect of oseltamivir on lower respiratory tract complications on a 2003 meta-analysis of ten randomized controlled trials (RCTs) by Kaiser and colleagues, but only two of the trials were published in the peer-reviewed literature.44–46 Hayashi requested the Cochrane group analyze all the data independently. The Cochrane group promptly attempted to do this, but what followed was nothing short of a farce.44,47–60 Roche, who manufactures oseltamivir, stymied the authors from obtaining the data from the eight unpublished RCTs, and because the Cochrane authors were unable to obtain the data from the eight unpublished RCTs, they accordingly changed their conclusion in their updated review, which per Cochrane rules was due six months after Hayashi’s critique.44,47–51 During their continued pursuit of the trial data, the Cochrane authors also came to realize that they would have to dig far deeper than they had anticipated, ultimately resulting in them requesting the original full clinical study reports (CSRs) from Roche; the plight is chronicled in great detail in The BMJ, the Cochrane reviews, and PLoS Medicine.52–60 The BMJ’s website also has extensive details, including a timeline and catalogued correspondence.44,47,48 Fiona Godlee (Editor-in-Chief of The BMJ), Tom Jefferson and Peter Doshi (two authors of the Cochrane review, with Jefferson as lead author), and others fiercely pursued the issue. As a result of this endeavor, The BMJ also ultimately started the “Open Data” campaign on October 29, 2012, with the first campaign being the “Tamiflu campaign.”47,48
In late 2013, Roche finally made 77 minimally-redacted CSRs available to Jefferson and his colleagues, and they were subsequently able to undertake an unprecedented review of CSRs that required herculean effort. Their updated review on the effects of oseltamivir in healthy adults and children was finally published in 2014 (their designation of “previously healthy” allowed chronic illnesses such as asthma, diabetes, or hypertension, but did not allow conditions with greater potential to impact immune system functioning, such as malignancy or HIV).59 The reviewers ultimately assessed 23 oseltamivir trials in Stage 1 of the review (where Jefferson and colleagues assessed for appropriate trial design and reliability and completeness of data); as a result, three of these 23 trials were ultimately excluded from Stage 2 of the review (the full analysis following Cochrane standards). Stage 2 included 20 trials amounting to 9,623 patients in total (6,574 in treatment trials and 3,049 in prophylaxis trials): 11 trials were on the treatment of adults, four were on the treatment of children, two were on prophylaxis in adults, two were on prophylaxis of the elderly, and one was on prophylaxis in households. The results are not terribly flattering for oseltamivir.59
To give a brief overview of the results, oseltamivir was found to reduce symptom duration by about 16.8 hours in adults (95% CI, 8.4 – 25.1 hours; P < 0.0001), amounting to roughly 0.70 days (95% CI, 0.35 – 1.05 days). In children, no beneficial effect was noted overall. When stratifying children by presence of asthma, children with asthma did not benefit, but otherwise healthy children experienced a reduction in symptom duration by about 29.4 hours (95% CI, 11.8 – 47.0 hours; P = 0.0011), amounting to roughly 1.21 days (95% CI, 0.49 – 1.96 days); however, the results in healthy children are based on a single trial with 669 participants, and the results in children with asthma are based on two trials with a total of 660 participants between the two trials.
The authors found no beneficial effect of oseltamivir on hospitalizations, serious complications (pneumonia was considered individually and will be discussed below), bronchitis, sinusitis, or otitis media.
Pneumonia was considered as an individual outcome. Noteworthy issues with pneumonia as an endpoint included it being a self-reported, investigator-mediated, unverified diagnosis, and the documentation was at times weakly regimented. However, with this understanding, the authors found an absolute risk difference (ARD) of 1.00% in adults (95% CI, 0.22% – 1.49%; risk ratio [RR], 0.55; 95% CI, 0.33 – 0.90; P = 0.017), which means 100 adults would have to be treated with oseltamivir to prevent one case of pneumonia (number needed to treat to benefit one person [NNTB], 100; 95% CI, 67 – 451). However, in the analysis limited to more detailed documentation of pneumonia (which had just over one-third the amount of patient data that the less specific documentation method had), no statistically significant benefit was found for prevention of pneumonia. No study reported the effects of oseltamivir on pneumonia confirmed by radiography (a rather notable shortcoming of the available trial data), and there was no apparent effect on pneumonia in children whatsoever.
For adult prophylaxis, the review found that oseltamivir helped reduce symptomatic influenza with an NNTB of 33 (95% CI, 26 to 55; ARD, 3.05%; 95% CI, 1.83% – 3.88%; RR, 0.45; 95% CI, 0.30 – 0.67; P = 0.000091). Effects of prophylaxis in children are not reported, as no trials of prophylaxis in children were included in Stage 2.
Adverse effects varied between active treatment and prophylaxis. If taking active treatment, nausea occurred significantly more often in adults with a number needed to treat to harm one person (NNTH) of 28 (95% CI, 14 – 112; ARD, 3.66%; 95% CI, 0.90% – 7.39%; RR, 1.57; 95% CI, 1.14 – 2.15; P = 0.0051); vomiting also occurred significantly more in adults with an NNTH of 22 (95% CI, 14 – 42; ARD, 4.56%; 95% CI, 2.39% – 7.58%; RR, 2.43; 95% CI, 1.75 – 3.38; P < 0.00001). In children, nausea was not significantly more common, but vomiting was with an NNTH of 19 (95% CI, 10 – 57; ARD, 5.34%; 95% CI, 1.75% – 10.29%; RR, 1.70; 95% CI, 1.23 – 2.35; P = 0.0014). If taking prophylaxis, headache (ARD, 3.15%; 95% CI, 0.88% – 5.78%; NNTH, 32; 95% CI, 18 – 115; RR, 1.18; 95% CI, 1.05 – 1.33; P = 0.0053) and nausea (ARD, 4.15%; 95% CI, 0.86% – 9.51%; NNTH, 25; 95% CI, 11 – 116; RR, 1.96; 95% CI, 1.20 – 3.20; P = 0.0068) both occurred significantly more often in adults while taking the prophylactic regimen. Psychiatric side effects while on prophylactic treatment approached significance (P = 0.062) and crossed the threshold of significance when the authors pooled on- and off-treatment events with an NNTH of 94 (95% CI, 36 – 1,538; ARD, 1.06%; 95% CI, 0.07% – 2.76%; RR, 1.80; 95% CI, 1.05 – 3.08; P = 0.031).
Most admirably, those wishing to scrutinize the data for themselves have open access to the entirety of the data.61 The full Cochrane review is now freely available (in 2013, the Cochrane Collaboration committed to making its reviews open-access one year after they are published), and an abridged version of the review has open-access availability as a separate article published in The BMJ.59,62
An individual-patient-data meta-analysis (IPD MA) of nine Roche-sponsored oseltamivir trials including 4,328 adult patients being treated for influenza was published in Lancet not too long after the updated Cochrane review was published.63 Although the methods are not as thorough or transparent as the Cochrane review, the primary outcome in the Lancet IPD MA was reduction in time to alleviation of symptoms, and for the full intention-to-treat (ITT) population, this outcome was a median of 17.8 hours (95% CI, 9.3 to 27.1 hours; time ratio, 0.85; 95% CI, 0.80 – 0.90; P < 0.0001), which is quite similar to the respective findings of the Cochrane review. In the analysis limited to patients with laboratory confirmation of influenza infection (PWCIIs), the reduction in time to alleviation of symptoms was 25.2 hours (95% CI, 16.0 to 36.2 hours; time ratio, 0.79; 95% CI, 0.74 – 0.85; P < 0.0001). Although this primary outcome was largely consistent with the findings of the Cochrane review, two other analyses – lower respiratory tract complications and hospitalizations – were emphasized by the study’s authors, and these findings were not consistent with those of the Cochrane review. In the analysis of PWCIIs, lower respiratory tract complications “requiring” antibiotics were lower among those receiving oseltamivir, with an ARD of 3.83% (95% CI, 2.18% – 5.05%; RR, 0.56; 95% CI, 0.42 – 0.75; P = 0.0001), translating to an NNTB of 27 (95% CI, 20 – 46). However, this was overwhelmingly due to cases of acute bronchitis (representing 81.71% of all the events in this outcome), a condition for which antibiotics are not indicated unless the etiology is Bordetella pertussis, which is uncommon.64–66 (Acute exacerbations of chronic bronchitis sometimes do require antibiotics, but this is not what the authors reference in their meta-analysis.) In the full ITT population, the ARD for lower respiratory tract complications was attenuated to 2.98% (95% CI, 1.65% – 4.00%; RR, 0.62; 95% CI, 0.49 – 0.79; P = 0.0001), with a corresponding NNTB of 34 (95% CI, 25 – 61), and the majority of the events (79.76%) were once again cases of acute bronchitis.
The authors also reported a lower rate of pneumonia in the PWCIIs, with an ARD, 1.00% (95% CI, 0.27% – 1.35%; RR, 0.40; 95% CI, 0.19 – 0.84; P = 0.015) and corresponding NNTB of 101 (95% CI, 75 – 376; minor apparent discrepancies in NNTBs and ARDs are the effect of rounding the ARDs to two decimal places). In the full ITT population, the ARD was 1.13% (95% CI, 0.62% – 1.40%; RR, 0.34; 95% CI, 0.18 – 0.64; P = 0.0009), with a corresponding NNTB of 89 (95% CI, 72 – 163). However, cases of pneumonia were again based on “participant report and the investigator’s clinical judgment” without radiographic confirmation.63(p1731) Furthermore, given the issues surrounding “pneumonia” as an outcome in the trials in both this analysis and the Cochrane review, one also has to question the rigor with which acute bronchitis was assessed, and the same can be said for lower respiratory tract complications “requiring” antibiotics in general. Indeed, the Lancet analysis provides no clear definition for such outcomes, which were also based on “participant report and the investigator’s clinical judgment,” and there appear to have been no criteria to confirm a bacterial infection or the “need” for antibiotics.63(p1731)
The other analysis emphasized by the Lancet IPD MA was a 1.06% ARD (95% CI, 0.32% – 1.40%; RR, 0.37; 95% CI, 0.17 – 0.81; P = 0.013) for being admitted to the hospital for any reason, translating to an NNTB of 94 (95% CI, 72 – 312); however, the reasons for admission were reportedly too varied to detect any pattern. Here again, we seem to be lacking a desirable granularity and clarity (in spite of the IPD MA approach). In the full ITT population, there was no significant difference in hospitalization.
With respect to adverse effects from oseltamivir, the Lancet IPD MA also found increases in nausea in both the PWCIIs (ARD, 3.93%; 95% CI, 1.57% – 7.00%; NNTH, 25; 95% CI, 14 – 63; RR, 1.60; 95% CI, 1.24 – 2.07; P = 0.0003) and the full safety population (ARD, 3.69%; 95% CI, 1.79% – 6.09%; NNTH, 27; 95% CI, 16 – 56; RR, 1.60; 95% CI, 1.29 – 1.99; P < 0.0001). It also found increases in vomiting in both the PWCIIs (ARD, 6.31%; 95% CI, 3.50% – 10.29%; NNTH, 15; 95% CI, 9 – 28; RR, 3.00; 95% CI, 2.11 – 4.26; P < 0.0001) and the full safety population (ARD, 4.70%; 95% CI, 2.73% – 7.33%; NNTH, 21; 95% CI, 13 – 36; RR, 2.43; 95% CI, 1.83 – 3.23; P < 0.0001). This review noted no significant increase in neurological or psychiatric outcomes in either population. There was an increase in “injuries and poisoning” in the full safety population (ARD, 0.49%; 95% CI, 0.02% – 1.98%; NNTH, 202; 95% CI, 50 – 5,990; RR, 3.37; 95% CI, 1.08 – 10.47; P = 0.036), but this was based on 15 events in the oseltamivir recipients and 4 events in the placebo recipients.
IPD MA is a powerful analytical modality, and the Cochrane authors also requested individual patient data, but they never received it despite follow-up correspondence (see the “Rapid Responses” to the abridged version of the review published in The BMJ62 and the catalogued correspondence with Roche48). Even with individual patient data, the IPD MA published in Lancet finds only small and uncertain effects, and it is not as thorough or transparent as that of the Cochrane group with respect to methodology. Furthermore, as the Cochrane review authors note, analysis limited to PWCIIs is likely problematic, introducing bias (from demonstrated imbalances in the treatment groups when analyzing only PWCIIs) and limited generalizability (from variable availability of influenza tests, which themselves can suffer from variable accuracy).59 Additionally, one notes the Lancet IPD MA was, on the surface, funded by the independent Multiparty Group for Advice on Science (MUGAS) foundation (for example, this is the funding source reported in the abstract on PubMed); however, an unrestricted grant from Roche really funded the study. Although not transparently reflected on PubMed, this is admitted within the body of the meta-analysis (see the “Acknowledgements” section at the very end of the article and the “Role of the funding source” subsection at the end of the “Methods” section). The authors claim Roche merely provided funding and had no other involvement, but this still introduces an unavoidable degree of uncertainty, and this is not ameliorated by discovering that two of the four authors have significant conflicts of interest (with one of the authors being on the Board of Directors of Gilead Sciences, which, although not specified in the disclosures, holds the patent on oseltamivir).
Although the current Cochrane review on oseltamivir is unprecedented in scale and even some of its methodology (i.e. reviewing individual CSRs), it represents a collective concerted effort to summarize the best-available research literature on oseltamivir in an unbiased manner, and it unequivocally represents the most robust, complete, and transparent assessment of the clinical trial evidence on oseltamivir.
Putting all this aside, the deeply troubling and more important observation – which is seen in the research providing an overview of these issues1–6 and which speaks to the core of this entire writing – is the utter lack of full publication and transparency. Hayashi made his critique in 2009. The updated Cochrane review could not be published until 2014 despite continual intensive efforts to gain access to the requisite data.
This is a problem.
Rather, it is a symptom, and the same symptoms are encapsulated in the aforementioned systematic reviews and other studies that collectively establish the indisputable and pervasive presence of these issues in the medical literature.1–6 These symptoms signify a grave illness in medicine, one that can only be corrected with a unified effort to provide the remedy.
The International Committee of Medical Journal Editors’ requirement of a priori clinical trial registration in order to be considered for publication has failed to provide this remedy.67–72 Similarly, ClinicalTrials.gov and the reporting mandate in Section 801 of the Food and Drug Administration Amendments Act of 2007 (FDAAA of 2007) have also failed, and the changes proposed in late 2014 by the National Institutes of Health (NIH) and the Department of Health and Human Services (HHS) unfortunately do not provide any material assurance of rectifying these issues.73–86 On the surface, the changes are desirable, as they call for an expansion in what exactly must be reported. Notably, however, the changes do nothing to address clinical trials that occurred before the FDAAA of 2007 initially went into effect; the changes are still reliant on the FDAAA of 2007, which has quite simply never been enforced; the changes would actually expand the responsibilities for enforcement even though there was a complete failure to enforce the FDAAA of 2007 in its original form; and the changes do not provide any explanation for the non-enforcement issue or any assurance of enforcement going forward.73–86
On the other hand, the AllTrials initiative (www.alltrials.net) – a joint initiative of (in alphabetic order): Bad Science, The BMJ, the Centre for Evidence-based Medicine, the Cochrane Collaboration, the James Lind Initiative, PLoS, and Sense About Science – has gained much momentum since its launch in 2013, and it continues to fight in this arena.87–89 It is being led in the U.S. by Dartmouth’s Geisel School of Medicine and the Dartmouth Institute for Health Policy and Clinical Practice, with the official launch in the U.S. occurring just this year in late July.87 Likewise, The BMJ remains very active in this arena, and Doshi and colleagues’ Restoring Invisible and Abandoned Trials (RIAT) initiative remains active as well.47,90–92 The Institute of Medicine (IOM) and World Health Organization (WHO) also recently made explicit calls to improve these issues.93,94 The remedy is long overdue, and we owe it to patients to come up with that remedy. Although this “case series” focuses heavily on industry (and with due cause), it must be noted that industry is not the only party at fault here; indeed, although not a novel finding, even the most recent appraisal of the failure of ClinicalTrials.gov and the FDAAA of 2007 shows that studies funded by governmental or academic institutions also suffer greatly from publication bias.82 Importantly, the AllTrials initiative, the activities of The BMJ, the RIAT initiative, and the calls of the IOM and WHO are not simply glittering generalities that ultimately lack substance – they all make concerted efforts to meaningfully address the issues, and contained within these efforts are suggested remedies. However, in order for these issues to be resolved, we still desperately need a unified voice from those within the scientific and medical community, and those outside the medical and scientific community should be a part of this as well. Publication bias and inadequate research transparency are scientifically and medically reprehensible and in violation of the basic ethical principles set forth in the World Medical Association’s Declaration of Helsinki.95 These issues have existed and gone unfettered for far too long. It is time to change that.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)