Keywords
microbiota, metabolism, diet
microbiota, metabolism, diet
It has long been appreciated that the Western diet—high in simple carbohydrates, processed meat, and fat—is associated with adverse health outcomes, including obesity, metabolic syndrome, and type 2 diabetes1,2. In particular, consumption of saturated fatty acids and industrially produced trans fatty acids is linked with metabolic syndrome and obesity3. However, controversy continues to surround the relative importance of fat in the diet overall and which fats are healthier or more harmful4. Saturated fat in diet has received much attention for its ability to induce chronic low-grade inflammation, widely recognized as a key link to the pathologies of obesity, type 2 diabetes, and cardiovascular disease5. Dietary fat drives chronic low-grade inflammation by expanding white adipose tissue (WAT), promoting macrophage recruitment to WAT, and generating adipose inflammation (reviewed in 6). Increased release of fatty acids from expanded WAT results in decreased muscle cell surface expression of the glucose transport protein GLUT4, reducing insulin-stimulated glucose uptake and inhibiting glycogen synthesis7. Impaired glucose uptake by GLUT4 is a key feature of insulin resistance (IR), which is a precursor to the development of type 2 diabetes.
Impairment of insulin action and inflammation from dietary fat have been described as resulting from the body’s limited capacity to store energy as fat8. In this view, dietary energy intake in excess of adipose storage capacity causes ectopic fat deposition in non-adipose tissues. Obesity and ectopic fat, in turn, are associated with muscle and liver accumulation of diacylglycerol (DAG)9 and ceramide, a sphingolipid derived from saturated fatty acids such as palmitate10. Toxic lipid molecules, generated through de novo synthesis from dietary fat, have pleiotropic effects on metabolism (reviewed in 11). DAG and ceramide have been shown to impair mitochondrial function, inhibit insulin signaling by acting on peroxisome proliferator-activated receptors (PPARs) and protein kinases, and cause inflammation via the nuclear transcription factor nuclear factor-kappa-B (NF-κB)9,10,12.
Despite equal energy content, dietary fatty acids that differ in structure can have opposite effects on inflammation and IR. The divergent fatty acid effects on metabolism cast doubt on a simplistic view of IR as a problem of limited adipose storage capacity. For example, saturated fatty acids but not polyunsaturated fatty acids (PUFAs) caused IR in Sprague-Dawley rats, although both dietary fats resulted in increased plasma-free fatty acids13. Similarly, incubation with saturated fatty acids palmitate and stearate caused IR in human skeletal muscle, whereas unsaturated oleate had opposing effects on insulin action14. In two human trials, substituting dietary saturated fat with polyunsaturated fat or monounsaturated fat improved insulin sensitivity and reduced visceral adiposity15,16. Certain dietary fats reduce adipose inflammation and IR, even in the overfed state17,18.
To understand why fats often have opposing metabolic effects, we note that the gut microbiota, the collection of microorganisms that inhabit our bodies and outnumber human cells by an order of magnitude, is sensitive to dietary composition and is linked to changes in metabolism and obesity19,20. The composition of the diet and gut microbiota interact to modify the risk of many chronic inflammatory diseases, including obesity, diabetes, and inflammatory bowel disease21. The metabolic responses to various fats might be best understood in light of dietary fat’s ability to drive changes in the makeup and function of the intestinal microbiota.
Recent studies have highlighted the central role of the gut microbiota in generating inflammation and regulating obesity and metabolism19. The microbiota consists of the collection of microbes living in and on our bodies, numbering as many as 100 trillion that reside mostly in the lower intestine22. Advances in sequencing technology and metagenomics have vastly increased the ability to identify intestinal microbes associated with obesity23 as well as mechanisms implicating microbiota in weight gain, such as increased energy harvest24. Compared with germ-free animals, conventionally raised mice have 60% more body fat even as the food intake was less25. This finding was explained by the suppression by gut microbes of the expression of a host intestinal protein known as fasting-induced adipocyte factor (FIAF). Because FIAF is an inhibitor of lipoprotein lipase (LPL), in the presence of gut microbes, less FIAF means reduced inhibition of LPL, resulting in more LPL, the enzyme responsible for importing and storing triglycerides. In a germ-free animal, greater FIAF increased the expression of genes responsible for fatty acid oxidation via stimulation of PPAR-γ coactivator and AMP-activated protein kinase26. Experiments that transferred microbes from obese and lean donors to germ-free mice support a causal role for microbiota in regulating fat mass and metabolism27. In a recent study, fecal microbiota from identical twins discordant for obesity were inoculated into germ-free mice, resulting in the transfer of the obese or lean phenotype of their donors28. Interestingly, when the resulting obese and lean mice were co-housed, the microbiota in lean mice appeared to have a selective advantage, transforming the microbiota of co-housed obese mice and causing weight loss. However, when obese mice were fed a high-fat diet, they could not be “rescued” by co-housing them with their lean counterparts28. In this example and others, an interaction of high-fat diet and specific microbiome appears necessary to cause systemic inflammation28,29.
A diet high in fat is sufficient to induce obesity and IR in many animal models4,30–33 and is associated with changes in gut microbiota and intestinal permeability. Two markers of inflammation, tumor necrosis factor-alpha and NF-κB activation, were induced in C57BL/6 mice fed a high-fat diet34. The essential role of the gut microbiota in this response was demonstrated by the absence of this effect in germ-free mice fed the same high-fat diet34. Because inflammatory markers increased before diet-induced obesity, inflammation that follows a high-fat diet may have a causal role in obesity34.
Highlighting the potent effects of dietary fat, a single high-fat meal was sufficient to induce pro-inflammatory signaling and IR35,36. IR and inflammation following a high-fat meal resulted from increased intestinal permeability to endotoxin35,36. In addition, because lipid A, the insoluble fraction of the endotoxin lipopolysaccharide, could be carried into the lymphatic system by chylomicrons, a high-fat meal could promote postprandial entry of endotoxin into the circulation even in the absence of increased intestinal permeability. In male C57BL6/J mice, high-fat feeding resulted in weight gain and a two- to three-fold increase in circulating endotoxin, a condition termed metabolic endotoxemia4. Weight gain and IR were equivalent in the group fed a high-fat diet and mice receiving a subcutaneous infusion of endotoxin4. From these results, it was proposed that the Western diet, high in fat and low in fiber, causes a dysbiosis that results in the translocation of gut-derived bacterial endotoxin37. Supporting the role of gut microbiota in this process, IR and weight gain were blocked with antibiotic pre-treatment38. IR by this mechanism involves endotoxin detection by the Toll-like receptor TLR4 and downstream pro-inflammatory signaling39–41. Recently, Everard et al. showed that the metabolic effects of high-fat diet require MyD88 (myeloid differentiation primary response gene 88), a central adaptor molecule for many TLRs with a key role in regulating inflammation and metabolism41. Mice with the MyD88 deletion were protected against high fat-induced metabolic endotoxemia and had increased regulatory T cells, findings that were linked with decreased IR and inflammation41. Additionally, MyD88 deletion altered the composition of the gut microbiota; transfer of those microbes into germ-free mice protected the recipient mice from high fat-induced IR. These results suggest that bi-directional control involving microbiota and the MyD88 pathway regulates metabolism and inflammation.
In this and following sections, we review how specific dietary fats alter the microbiome and change insulin sensitivity. Saturated fatty acids have been shown to have direct stimulatory effects on TLR expression42 and Jun N-terminal kinase (JNK) activity43 promoting IR via mechanisms independent of the gut microbiota. However, these direct effects may be less consequential than the influence of the gut microbiota on host metabolism, as underscored by the finding that germ-free animals are protected from high-fat diet-induced obesity and IR25,34. Saturated fatty acids have been shown to cause dysbiosis and intestinal inflammation in interleukin-10−/− mice by encouraging overgrowth of a bile-tolerant Gram-negative bacteria, Bilophila wadsworthia44. In another study of C57BL/6J mice, a diet high in saturated fat caused increased growth of three types of sulfidogenic bacteria, primarily in colonic mucosa45; these bacteria produce hydrogen sulfide gas as a metabolic by-product which can damage the intestinal barrier and cause endotoxemia. Feeding C57BL/6 mice a diet high in saturated fat decreased expression of tight junction proteins, causing increased intestinal permeability, endotoxemia46, and elevated lipopolysaccharide-binding protein47. In addition to higher fecal and plasma endotoxin levels, mice fed a diet high in saturated fat had fewer Bifidobacteria and increased Enterobacteriaceae in fecal culture46. Laugerette et al. showed an increased intestinal Escherichia coli population along with elevated plasma and adipose inflammation in animals fed saturated fat (palm oil) compared with unsaturated fats47. Taken together, these results support the hypothesis that certain diets high in saturated fatty acids may modify the structure and function of the gut microbiota, causing inflammation and IR in animal models (Figure 1).
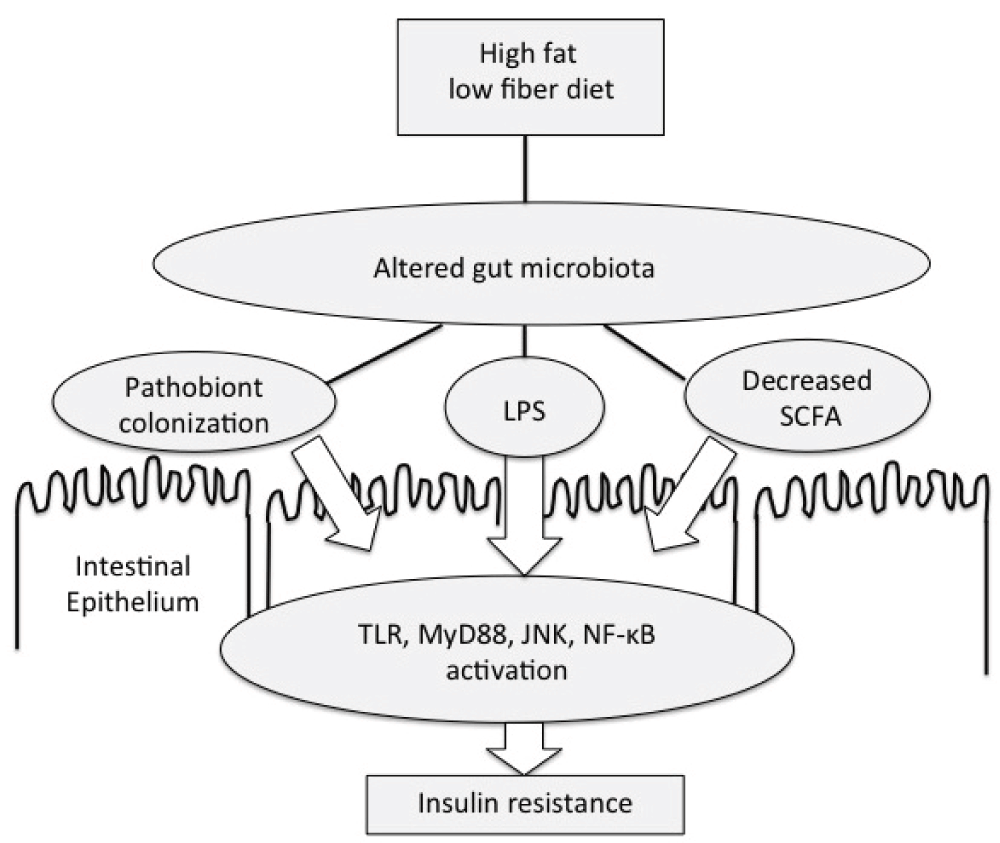
Gut microbiota play a central role in the metabolic endotoxemia model of obesity and insulin resistance. Diets high in fat and low in fiber alter the function and composition of the gut microbiota. These changes can increase systemic lipopolysaccharide (LPS) exposure, thereby contributing to low-grade inflammation and impairing insulin-stimulated glucose uptake by muscle. JNK, Jun N-terminal kinase; MyD88, myeloid differentiation primary response gene 88; NF-κB, nuclear factor-kappa-B; SCFA, short-chain fatty acid; TLR, Toll-like receptor.
Ghosh et al.48,49 showed that C57BL/6 mice fed a diet rich in omega-6 (n-6) PUFAs (corn oil) resulted in bacterial overgrowth and dysbiosis. The high n-6 PUFA diet, alone among the high-fat diets studied, was also associated with bacterial invasion of the intestinal epithelial cell layer48. Corn oil supplementation caused decreased spontaneous locomotor activity, hyperinsulinemia, and IR in female C57BL/6 mice50. This animal study provides an interesting insight into the “couch potato” sedentary state in humans, suggesting that diet and microbiota can influence voluntary physical activity.
Dietary n-6 PUFAs were linked with changes in the composition of the gut microbiota in C57BL/6 mice49. These changes included increased abundance of Enterobacteriaceae and segmented filamentous bacteria, bacterial groups associated with inflammation49. N-6 PUFA feeding to C57BL/6 mice was shown to increase the numbers of intestinal Proteobacteria51 and change gut microbiota composition along with weight gain and fatty infiltration of the liver52. Huang et al. also reported an increase in intestinal Proteobacteria after n-6 PUFA feeding in C57BL/6 mice and greater macrophage infiltration of adipose than observed with saturated fat diets53. Excess dietary N-6 PUFAs caused higher adipose expression of resistin, a hormone linked with inflammation and IR, than was observed after consumption of saturated fat53.
Ghosh et al. demonstrated that altered gut microbiota caused by n-6 PUFAs in 2-year-old C57BL/6 mice was prevented when omega-3 (n-3) PUFAs (fish oil rich in DHA and EPA) were added to the diet48, suggesting that n-3 PUFAs can protect against dysbiosis. N-3 EPA and DHA reversed bacterial overgrowth and reduced fatty diet-induced inflammation by recruiting regulatory T cells to the small intestine48. However, Mujico et al. showed no similar protection from n-3 PUFA supplementation from dysbiosis caused by saturated fatty acids18. Another recent study showed that mice fed fish oil had decreased abundance of Helicobacter and Pseudomonas and Firmicutes, organisms associated with ulcers, infection, and weight gain, respectively54. One mechanism that may account for dietary n-3 PUFA’s reduction of Helicobacter and Pseudomonas is that those organisms are sensitive to the direct bactericidal effects of EPA and DHA55,56. Bacterial killing by n-3 PUFAs and other fatty acids is likely important to the overall composition of the microbiota and the function of the intestinal barrier57,58.
Dietary fish oil strengthened intestinal barrier function and reduced plasma endotoxin levels in swine17. Fish oil has also been linked with reduced TLR activation and MyD88 signaling in swine59. In addition to having beneficial effects on metabolic endotoxemia, n-3 PUFAs were shown to stimulate the G-protein coupled fatty acid receptor GPR120, promoting insulin sensitivity by increasing cell surface expression of GLUT460,61. N-3 PUFAs have additional anti-diabetic effects by activating GPR 40, causing increased insulin secretion from pancreatic β cells.
Mujico et al. reported that oleic acid (a monounsaturated fatty acid) prevented high-fat diet dysbiosis in female ICR mice and increased the abundance of intestinal Bifidobacteria, a group associated with improved intestinal barrier function18. Oleic acid supplementation prevented weight gain and restored the proportion of microbial phyla altered by a high-fat diet18. Hidalgo et al. showed that butter produced changes in murine gut microbiota similar to those found in obese humans but that olive oil prevented those changes62. Interestingly, virgin olive oil had different effects on the microbiota compared with refined olive oil, suggesting that the non-lipid phenolic components of olive oil may account for some of its benefits62. Dietary supplementation with monounsaturated oleic acid in young adults improved insulin sensitivity, an effect not seen with saturated fat63. Downstream effects of microbiota may be responsible in part for improved insulin sensitivity and reduced type 2 diabetes observed with diets rich in olive oil and other monounsaturated fats64,65.
Short-chain fatty acids (SCFAs) are fully saturated but have fewer carbon atoms than long-chain saturated fatty acids, such as palmitate. SCFAs often have anti-inflammatory signaling properties (reviewed in 57). For instance, SCFAs such as butyrate tend to reduce inflammation by activating the SCFA receptor GPR4366. GPR43 activation increased energy expenditure and decreased adipose tissue insulin sensitivity while increasing insulin sensitivity in muscle and liver in C57BL/6 mice66. GPR43-deficient mice were obese on normal diet, whereas mice overexpressing GPR43 remained lean on a high-fat diet66. SCFAs are also a by-product of microbial fermentation of indigestible carbohydrates that are termed prebiotics when given therapeutically to alter the microbiota. Prebiotic treatment increased butyrate production in Wistar rats and was associated with increased Bacteroidetes, whereas high-fat diet reduced formation of butyrate and increased liver fat and inflammation39. Butyrate has anti-obesity effects by stimulating the expression of angiopoietin-like protein-4 (ANGPTL4) in human epithelial cells, leading to reduced expression of LPL and increased lipolysis67. Thus, microbes that preferentially generate butyrate through fermentation have a favorable effect on metabolism.
Prebiotic treatment had additional metabolic benefits by increasing the abundance of Akkermansia muciniphila, a group of mucin-foraging bacteria that were depleted in obese and type 2 diabetic mice68. A. muciniphila separately prevented visceral adipose inflammation, increased anti-inflammatory regulatory T-cell numbers, and improved insulin sensitivity in C57BL/6 mice69. Improvement of glucose tolerance in db/db fiber-fed mice was recently shown to be transmissible with fecal transplantation, even when the recipient mice were never exposed to dietary fiber70. The insulin-sensitizing effects of dietary fiber in donor and recipient mice in that study were attributed to increased Lactobacillus and Bifidobacterium, decreased Alistipes, and changes in amino acid fermentation70. These findings underscore the importance of fiber-fermenting gut bacteria in regulating insulin sensitivity by the action of SCFAs and because of changes in gut microbiota function.
The studies in this review showing a central role of the microbiota in regulating metabolism and immune function challenge traditional concepts of lipotoxicity as a primary cause of IR and metabolic syndrome8. Explanations of metabolic diseases that center on toxic lipid mediators from overfilled adipose depots are inadequate to explain the widely variable effects of equicaloric fats that have been reviewed here and elsewhere (for example, 71). A more unifying explanation is that the metabolic changes, inflammation, and changes in fat storage leading to obesity are outcomes of dietary fat acting on both the host and the gut microbiota as well as of diet-driven crosstalk between the host and the microbiota (Figure 2). Protection from obesity and IR in the germ-free state25 and with antibiotic treatment38 provides strong support for this view, which is a departure from the traditional understanding of how metabolic disorders are caused by dietary fat.
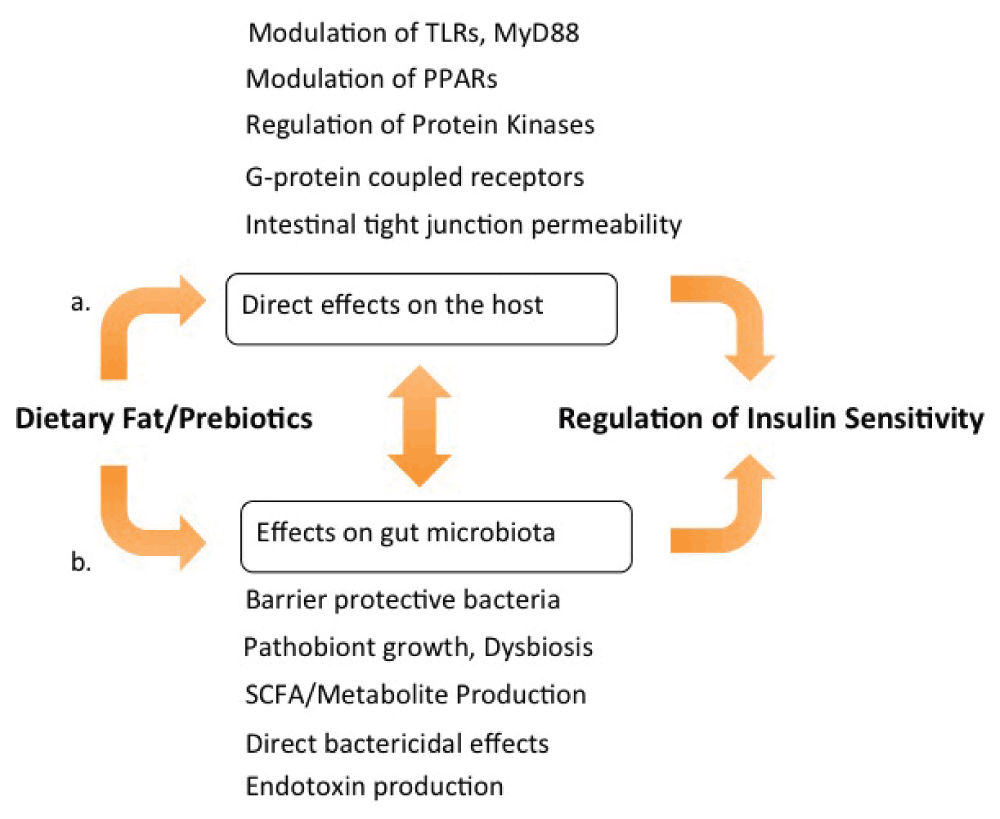
(a) Dietary fats directly stimulate G protein-coupled receptor (GPR) fatty acid receptors and alter intestinal tight junction protein expression, also affecting the expression and activity of Toll-like receptors (TLRs) and the adaptor protein MyD88 (myeloid differentiation primary response gene 88), modulating nuclear transcription factor activity, and regulating inflammation and metabolism. (b) Dietary fats and prebiotics impact the composition and function of the gut microbiota and affect intestinal permeability. Significant cross-talk occurs between microbiota-derived signals, such as endotoxin and short-chain fatty acid (SCFA), and the pathways described in (a). Cues from both sources, diet and microbiota, are integrated to modulate inflammation and insulin sensitivity. PPAR, peroxisome proliferator-activated receptor.
It has been suggested that microbiota-induced changes in metabolism can be adaptive for the mammalian host (for instance, by diverting energy to fetal growth during pregnancy)72. The general concordance between insulin sensitivity/resistance and fat-driven changes in the microbiome described in this review (Table 1) suggests an alternative evolutionary explanation. Specifically, nutrients may serve a signaling function to the immune system in mammals by conveying information about diet-driven changes in the gut microbiota57. This hypothesis makes two predictions: (1) nutrients that lead to dysbiosis may generate pro-inflammatory signaling, and (2) nutrients that prevent dysbiosis may trigger anti-inflammatory signaling. A review of the effects of dietary fats on inflammation and gut microbes tended to be in line with these predictions57. As suggested by the present review, the metabolic effects of dietary fats can often be predicted by their effects on the microbiome, perhaps because metabolism and inflammation share similar regulatory pathways. We further propose that the modulation of metabolism by fats and microbiota may be adaptive in fueling the increased energy needs of immune cells activated by dysbiosis. By blocking glucose uptake, IR reduces energy utilization by tissues dependent on GLUT4 glucose uptake (predominantly skeletal muscle and fat) and diverts energy access to tissues not reliant on insulin-stimulated GLUT473–75. Phagocytes (for example, macrophages) and intestinal epithelial cells do not rely on GLUT4. As a result, glucose energy is expected to be preferentially delivered to activated innate immune cells in the gut during the IR state.
| Dietary fat | Example (structure) dietary source | Effect on gut microbiota** | Effect on intestinal barrier | Insulin resistance | Inflammation |
|---|---|---|---|---|---|
| Saturated fatty acid | Palmitic acid (16:0)* Dairy | Enterobacteriaceae dysbiosis Decreased Bifidobacteria Increased ratio of Firmicutes/ Bacteroidetes [18,44,53,68] | Increased endotoxemia [17,35] | Increased [68] | Increased [30,35] |
| N-6 PUFA | Linoleic acid (18:2) Corn oil | Enterobacteriaceae dysbiosis [48] | Proteobacteria translocation [49] | Increased [78] | Increased [49,50] |
| N-3 PUFA | Docosahexaenoic acid (22:3) Marine fish | Prevented n-6 dysbiosis [49] | Prevented Proteobacteria translocation, endotoxemia [17,49] | Reduced [60] | Reduced [49] |
| Monounsaturated fatty acid | Oleic acid (18:1) Olive oil | Prevented saturated fat dysbiosis [18] | No change [17] | Reduced [65] | Reduced [63] |
| SCFA | Butyrate (4:0) Dietary fiber | Normalized Firmicutes/ Bacteroidetes [68] | Decreased permeability (with prebiotics) [39] | Reduced [68] | Reduced [66] |
Saturated fats have been reported to cause dysbiosis and have been linked in animal studies with increased Enterobacteriaceae, increased Firmicutes/Bacteroidetes ratio, and decreased Bifidobacteria, among other changes. Dietary omega-6 (n-6) polyunsaturated fatty acid (PUFA) also is reported to cause an Enterobacteriaceae dysbiosis in mice. These microbiota alterations are associated with increased intestinal permeability, insulin resistance, and inflammation. Dysbiosis caused by saturated fat and n-6 PUFA was reversed with supplementation with monounsaturated oleic acid, omega-3 (n-3) PUFA, and short-chain fatty acid (SCFA) precursors (prebiotics). Protection from dysbiosis with oleic acid, n-3 PUFA, and prebiotic supplementation is accompanied by decreased inflammation and increased insulin sensitivity. These patterns are consistent with increased insulin-independent glucose uptake by the activated immune system during dysbiosis and opposite shifts in energy utilization when dysbiosis is absent.
Despite murine studies suggesting dysbiosis, inflammation, and metabolic disease from n-6 PUFAs, some human studies have shown no harm, and possible benefit, from consuming n-6 fats76. A recent longitudinal cohort study in Finland showed reduced risk of metabolic syndrome with increased n-6-to-n-3 PUFA ratio in serum77. Although molecular and animal studies imply a therapeutic benefit of n-3 PUFAs for metabolic syndrome and diabetes, observational studies of n-3 fat and type 2 diabetes have been mixed, indicating a possible reduction of type 2 diabetes risk with fish consumption in Asian populations78 but no benefit from fish consumption in a recent European case control study79. Elevated circulating n-3 fatty acids were recently linked to increased insulin sensitivity in overweight men80 and randomized trials have shown improved parameters related to metabolic syndrome with n-3 PUFA supplementation, including increased adiponectin and improved triglyceride levels in overweight women81,82. Self-reported diets high in n-3 alpha-linolenic acid, n-6 linoleic acid, and monounsaturated oleic acid have been associated with improved glucose metabolism76. Taken together, these findings indicate possible protection from metabolic syndrome from n-3 PUFAs and support the idea that unsaturated fats are more metabolically healthy than saturated fats. However, a recent study challenged the concept that saturated fats are harmful83. Unlike experiments in which milk fat caused dysbiosis and inflammation in mice44, experiments in humans given a diet high in dairy fat showed no increase in inflammation83. Moreover, improvement in insulin sensitivity and reduced adipose fat occurred more in human subjects assigned a diet low in carbohydrates rather than a diet low in saturated fat84. These results suggest that weight loss and improvements in metabolism can result from diets that prioritize reduction of carbohydrates rather than fats.
One explanation for the disparities between human and animal studies is that people are not mice85 and murine models may be poorly suited to understand human metabolism. Mammalian-microbiota co-evolutionary history is different for humans and other animals86, and a likely consequence is that foods modify the human microbiome differently and have distinct regulatory effects on immunity and metabolism. To date, we cannot define exactly what those species-level differences are. Without that information, it is too early to give a blanket recommendation for or against any class of dietary fat, especially when specific fatty acids in the same class vary in effects and depend also on an individual’s unique microbiota and genetic background. Molecular and human epidemiologic data strongly indicate that some fats are better for metabolic health than others. More human and comparative studies will be needed to determine whether metabolically healthy fats are those that maintain a healthy microbiota.
DAG, diacylglycerol; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; FIAF, fasting-induced adipose factor; GLUT-4, glucose transporter type 4; GPR, G protein-coupled receptor; IR, insulin resistance; LPL, lipoprotein lipase; MyD88, myeloid differentiation primary response gene 88; n-3, omega-3; n-6, omega-6; NF-κB, nuclear factor-kappa-B; PPAR, peroxisome proliferator-activated receptor; PUFA, polyunsaturated fatty acid; SCFA, short-chain fatty acid; TLR, Toll-like receptor; WAT, white adipose tissue.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)