Keywords
Primary microcephaly, Delayed development, Periventricular leukomalacia, White matter hyperintensities, Diabetes mellitus, TRMT10A, tRNA methyltransferase
This article is included in the Rare diseases collection.
Primary microcephaly, Delayed development, Periventricular leukomalacia, White matter hyperintensities, Diabetes mellitus, TRMT10A, tRNA methyltransferase
We present case histories of siblings (a sister and brother) born to healthy non-consanguineous parents, with microcephaly and delayed development.
The first patient is a 12 year old girl who was born at 39 weeks gestation weighing 4 lb 10 oz, without perinatal difficulties. She was healthy as an infant, except for recurrent ear infections, which were treated with tympanostomy tubes. Hypotonia was noted during infancy, but she reached her early motor milestones, including crawling and walking, at the normal ages. Speech delay and behavioral problems were noted by 4 years of age. There was no regression. Ophthalmological evaluation was normal. Physical exam at age 4.5 years showed microcephaly, low anterior hair line, deep set eyes with mild hypotelorism, shortened forehead, and her neurological exam was normal. Head circumference was 3.5 SD below the mean for age in girls. These features have remained constant at subsequent examinations.
MRI scan of the brain showed mild FLAIR/T2 hyperintensities in the periatrial white matter (Figure 1A); a CT scan did not show calcifications. Skeletal x-rays did not show any abnormality of her bones. Hearing tests, renal ultrasound, and echocardiogram were all normal. High resolution karyotype and array comparative genome hybridization (CGH) were normal. Carbohydrate deficient transferrin study, lysosomal enzymes, and 7-dehydrocholesterol level were also normal. She has not had documented hypoglycemia or hyperglycemia. Psychometric testing led to the diagnosis of mild intellectual disability. Speech and occupational therapy were provided through school and she has continued to make steady developmental progress.
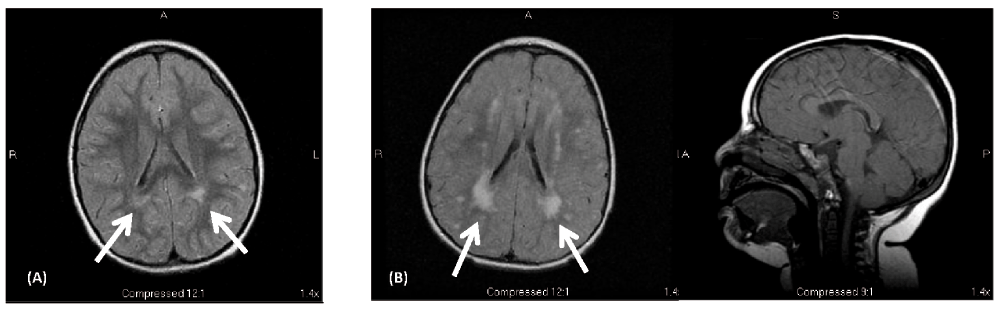
(1-A) – Axial MRI FLAIR image from the affected female child done at 4.5 years of age shows hyperintensities in the periventricular (primarily periatrial) white matter (arrows). (1-B) Axial FLAIR and sagittal T1 MRI images from the affected male child at 2.5 years of age. This shows bilateral periventricular (periatrial) and subcortical white matter hyperintensities consistent with dysmyelination (arrows). Brain architecture (corpus callosum, cerebellum, and cortical gyration) was normal.
The second patient is a 10 year old boy who was born at 36 weeks gestation weighing 5 pounds, and developed mild neonatal jaundice. The pregnancy was complicated by oligohydramnios and decreased fetal movement. He was diagnosed with hypotonia during infancy. He too achieved motor milestones at appropriate times (walked at 15 months) but speech was delayed. He had an episode of febrile status epilepticus at 17 months, and subsequent epileptic seizures without fever. Seizures have been controlled with levetiracetam (50 mg/kg/day, corresponding to 500 mg twice daily). A recent EEG showed symmetric, bilateral, frontally dominant polyspike and wave discharges, consistent with generalized epilepsy. He has a history of recurrent pulmonary infections and reactive airway disease; initial testing by CFTR gene sequencing was not conclusive and revealed only benign variants. Sweat chloride testing was abnormal on one occasion and then borderline, pancreatic elastase was normal, and a clinical diagnosis of cystic fibrosis was made. He also had recurrent otitis media that required tympanostomy tubes.
Physical examination at age 2.5 years showed small stature (5% for height and weight) microcephaly (head circumference 3 SD below mean for age), low anterior hair line, deep set eyes with mild hypotelorism, and a normal neurological exam. His phenotype was similar to his older sister, although he seemed to be more severely affected. MRI of the brain at age 2.5 years showed extensive FLAIR/T2 hyperintensities in the periventricular and subcortical white matter (Figure 1B).
The following biochemical and genetic tests were normal: array CGH, fragile X test, TORCH titers, quantitative plasma amino acids, quantitative urine organic acids, fluorescent in-situ hybridization (FISH) test for Pelizaeus-Merzbacher disease, very long chain fatty acids, enzyme assays for CLN1 (PPT1 – palmitoyl-protein thioesterase 1) and CLN2 (TPP1-tripeptidyl-peptidase 1), and 7-dehydrocholesterol. He has not had documented hypoglycemia or hyperglycemia.
His features have remained constant on follow-up examinations, with persistent microcephaly and mild hypotelorism. He is making slow progress in school with speech, occupational and physical therapy.
Patients and parents were enrolled into a clinical research protocol sponsored by the Translational Genomics Research Institute (TGen) approved by the Western Institutional Review Board, Protocol Number 20120789. After informed consent, blood samples were collected for DNA and RNA extraction. A separate buccal swab was sent for Sanger sequencing and independent confirmation of selected variants in a clinical laboratory (GeneDx, Gaithersberg, MD).
Libraries were prepared using the Illumina’s TruSeq DNA sample preparation kit and the TruSeq exome enrichment kit (Illumina, Inc., San Diego, CA, USA), following the manufacturer’s protocol. Sequencing was done by 100-bp paired-end sequencing on a HiSeq2000 instrument (Illumina, Inc., San Diego, CA, USA). Reads were aligned to the Human Genome (Hg19/GRC37) using Burrows-Wheeler transform alignment (BWA v.0.7.5)1. PCR duplicates were removed using Picard v.1.922, and base quality recalibration, indel realignment and single nucleotide polymorphism (SNP) and indel discovery were performed using the Genome Analysis Toolkit (GATK v.2.5-2)3. Variants were annotated with SnpEff 3.2a and selected (SnpSift) for protein-coding events. Prediction scores were loaded from dbNSFP and used for filtering.
Exome sequencing identified 33,640 SNPs and indels common to the two affected individuals. The majority of these variants were common in the population, and only 4,355 were considered either novel, private or rare variants. An annotated variant file was created that included variants in any of the four family members (male child, female child, father, and mother). Excluding UTR (untranslated region) and synonymous variants left 452 candidate variants.
There were no variants consistent with de novo mutation in both children (with presumed germline mosaicism in a parent). Four variants were identified consistent with a homozygous recessive model: SERINC2, PTH2R, SGK223, and PMP22. None of these were consistent with the clinical phenotype.
We considered an X-linked recessive model (female child less severe than male) and identified variants were in NHS (Nance-Horan syndrome), DDX53, WAS, and TREX2; four variants in RPGR. The TREX2 variant has been reported in 83 hemizygotes in the ExAC Browser database v 0.3 (Broad Institute). The DDX53 variant is rare and has been implicated as a potential X-linked autism locus. X chromosome inactivation studies in the carrier mother have not been done.
When we considered compound heterozygous model, we identified only two variants in a single gene, TRMT10A. Both children have inherited a maternal variant encoding a nonsense mutation, c.277C>T (p.Arg93*) and a paternal variant encoding a nonsense mutation, c.397C>T (p.Arg133*). The Arg133* variant has been observed in one of 120,000 alleles in the ExAC Browser database, while the Arg93* allele has never been reported before. Based on the damaging nature of these mutations (protein truncation), consistency with an autosomal recessive model, and two previous reports of homozygous TRMT10A mutations in children with primary microcephaly4,5, we concluded that the TRMT10A mutations were causal for our patients’ phenotype.
Microcephaly is a clinical condition, rather than a specific diagnosis, defined as having a small brain size, measured by head circumference. It is generally accepted that head circumference less than 2–4 standard deviations below the mean for age and gender is considered to be microcephaly6,7.
Primary microcephaly is either congenital and present at birth, or progressive and caused by postnatal decrease in head circumference growth rate (as in Rett syndrome, for instance). Congenital microcephaly occurs more frequently in patients born to consanguineous parents. Congenital microcephaly can be further classified by clinical and imaging features set forth by Barkovich and others8,9. Patients with autosomal recessive primary microcephaly (MCPH) have microcephaly at birth and non-progressive mental retardation10,11. Microcephaly with normal or simplified gyration (MSG) is thought to be a result of abnormal neuronal and glial proliferation or apoptosis12.
Many genes have been implicated in the development of microcephaly (summarized in Table 1) including ASPM, MCPH1, CDK5RAP2, CEP152, CENPJ, WDR62, STIL, CASC5, CEP135, ZNF335, PHC1, CDK6, with many of them contributing to centrosome and spindle protein dysfunction during mitosis6,10,13–17. Mutations in the ASPM gene are the most common in this group, and account for 30–40 percent of these cases18. Genes implicated in primary microcephaly with associated features such as cognitive and motor impairment and epilepsy include SLC25A19, ATR, ARFGEF2, and RAB3GAP119. PYCR2 mutation described by Nakayama causes dysfunctional proline synthesis and leaves affected patients more susceptible to oxidative stress induced apoptosis, leading to postnatal microcephaly from cell death in the developing brain20.
This table contains a summary of selected primary microcephaly syndromes, including MCPH 1–15. Information provided includes a brief description of the phenotype, gene that is mutated, and pathogenic mechanism.
Primary microcephaly is a result of a defect in the generation of the appropriate number of neurons during early neural development. Cell division of neural progenitors in the neuroepithelium can be symmetric (generating two progenitors) or asymmetric (generating one progenitor and one post-mitotic neuron). Microcephaly has been associated with microtubule, centrosome, and mitotic dysfunction. Dysfunction in the mitotic machinery may cause inappropriate asymmetric cell division during neurogenesis in the developing cortex and subventricular zone21,22. Abnormal control of apoptosis during the neurogenesis phase of development can also lead to microcephaly. Studies in mice have demonstrated microcephaly due to an increase in neuronal apoptosis and depletion of the neural progenitor population23. Specifically, endoplasmic reticulum stress-induced apoptosis has been shown to cause microcephaly and early onset diabetes from beta cell ER stress-induced apoptosis.
TRMT10A (also known as RG9MTD2 – RNA (guanine 9)-methyltransferase domain-containing protein 2) is homologous to a yeast tRNA methyltransferase (Trm10) and methylates tRNAs at the m1G9 position. TRMT10A is expressed in several tissues (liver, kidney, spleen, lung, fat) but at especially high levels in brain and pancreatic islet cells4. Mutation of another tRNA methyltransferases, NSUN2, has been associated with microcephaly, short stature, intellectual disability, and facial dysmorphism24,25.
There are two previous reports of patients with homozygous nonsense or missense mutation of TRMT10A resulting in microcephaly. In the first report, Igoillo-Esteve and colleagues described 3 siblings, from a large consanguineous Moroccan family, with microcephaly, intellectual disability, short stature, and early-onset diabetes4. Whole exome sequencing identified a homozygous p. Arg127* nonsense mutation in the TRMT10A gene in the 3 affected patients; both parents and an unaffected brother were heterozygous for this mutation. Features in the oldest patient (at age 26), in addition to microcephaly and intellectual disability, included short stature, absence seizures, development of diabetes at age 22, and a variety of dysmorphic features. Brain MRI showed normal architecture and gyration. The other two children developed diabetes at age 19 and 14 respectively. The TRMT10A Arg127* mutation resulted in greatly reduced mRNA levels (presumably by nonsense-mediated decay) and absent protein levels. Loss of TRMT10A protein induced apoptosis in rat β-cells and increased stress-induced apoptosis by many endogenous molecules including high levels of glucose4.
In a second report, Gillis and colleagues described 3 siblings (one female and two male) born to healthy, non-consanguineous parents from a small, inbred Jewish community of Uzbekistan5. All 3 patients suffered from microcephaly, intellectual disability, short stature, seizures, and altered glucose metabolism. The female child had delayed pubertal development and was amenorrheic at age 19 years. Exome sequencing in one affected patient followed by targeted genotyping in the rest of the family (parents, two affected sibs and 9 unaffected sibs) defined a homozygous p. Gly206Arg missense mutation in TRMT10A as causing disease in this family. All three affected patients had episodes of hypoglycemia, hyperinsulinemia, and abnormal response to glucose. Brain imaging in two of these patients was normal. The Gly206Arg mutation resulted in almost complete loss of methyltransferase activity5. This suggested that decrease in methylated tRNA is responsible for growth failure, developmental delay and impaired glucose metabolism.
TRMT10A microcephaly, in contrast to primary microcephaly linked to centrosome and spindle function during mitosis, is likely related to the loss of brain volume due to increased apoptosis. TRMT10A deficiency increases the amount of beta cell apoptosis, and presumably neuronal apoptosis. The development of diabetes and abnormal glucose response may be related to endoplasmic reticulum stress-induced apoptosis induced by endogenous fatty acids and high levels of glucose4. Endoplasmic reticulum stress-induced apoptosis has also been implicated in a syndrome of primary microcephaly associated with simplified gyral pattern, epilepsy, and infantile diabetes caused by mutation of the IER3IP1 gene19.
Here we report two additional patients with primary (congenital) microcephaly and intellectual disability caused by compound heterozygous nonsense mutations of TRMT10A. These patients have not displayed hypoglycemia nor early onset diabetes. Brain imaging did show abnormalities in the central white matter, suggesting delayed or abnormal myelination. TRMT10A is not currently included in microcephaly gene panels available from commercial laboratories, stressing the importance of exome sequencing in the genetic diagnosis of primary microcephaly. The correct genetic diagnosis in our patients allows for anticipation and early treatment of medical complications by institution of meticulous glucose monitoring and control from an early age.
Written informed consent for publication of their clinical details and clinical images was obtained from the parent of the patients.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)