Keywords
next generation sequencing, docker, container, variant caller, pipeline, read alignment, reproducible, bioinformatics
This article is included in the Container Virtualization in Bioinformatics collection.
next generation sequencing, docker, container, variant caller, pipeline, read alignment, reproducible, bioinformatics
Bioinformatic pipelines are frequently composed of large numbers of loosely coupled pieces of software, each tool requiring substantial configuration, maintenance and management of dependencies. Historically to facilitate packaging and reuse of pipelines, management frameworks such as Galaxy1, Ruffus2, and Taverna3 have been developed. While these workflow management systems work well, portability and deployment complexity limit their usability.
Our primary motivation for developing NGSeasy was to simplify pipeline deployment for academic and clinical labs, minimising the burden of informatic support. To achieve this, we used Docker4, an emerging container-based virtualization technology. Compared to virtual machines, Docker containers are simply a set of processes running in a multi-tenanted Linux host kernel, so are very lightweight as there is no underlying machine to emulate. These containers capture the initial investment of effort to build and configure them greatly facilitating re-use, they can be easily extended to modify or incorporate new components and shared on private or public (Docker Hub) registries.
Using NGSeasy and Docker, bioinformaticians and more importantly, researchers with fewer bioinformatic skills can very quickly deploy the pipeline to different environments e.g. development, testing and production, with the knowledge that the containers should always run consistently. Furthermore, we support multiple versions of the NGSeasy containers on Docker Hub, as each container packages its own dependencies and is versioned, the fidelity of the analysis is preserved in future execution – a requirement for reproducible research and clinical auditing5.
NGSeasy has provided us with the opportunity to start defining and thinking about best practices for building Dockerised modular pipelines. Many of these practices have been adapted in our images. Our (compbio/ngseasy-base) image forms the foundation layer on which each pipeline container application is built.
All Dockerfiles used to generate the NGSeasy images are available at https://github.com/KHP-Informatics/ngseasy.
We include what we think of as some of the best and most useful NGS "power tools" in compbio/ngseasy-base image (Table 1). These are all tools that allow the user to manipulate BED/SAM/BAM/VCF files in a variety of ways.
| Component | Description | Version |
|---|---|---|
| samtools6 | Parse [s/b]am | 1.2-17 |
| bcftools7 | Parse vcf | 1.2-5-g7fa0d25 |
| vcftools8 | Parse vcf | v0.1.12b |
| vcflib9 | Parse vcf | v1.0.0 |
| bamUtil10 | Parse [s/b]am | 1.0.13 |
| bedtools11 | Parse [s/b]am/bed | v2.23.0-10 |
| samblaster12 | Parse [s/b]am | 0.1.21 |
| sambamba13 | Parse [s/b]am | v0.5.1 |
| seqtk14 | Parse FASTQ | 1.0-r77 |
| vt15 | Parse VCF | *Latest |
| vawk16 | Awk-like VCF parser | 0.0.2 |
| bioawk17 | Awk-like NGS parser | *Latest |
Our feature rich base image, allows pipes and streamlined system calls for manipulating the output of NGS pipelines, namely, BED/SAM/BAM/VCF files. Therefore, we built these into a single development environment for NGSeasy. This image is used as the base for all of our compbio/ngseasy-* tools.
A more Docker-esque approach, would be to have separate containers for each NGS tool. However, this belies the fact that many of these tools are required to interact, e.g. through pipe calls, when used as part of a streamlined pipeline.
Many of the raw NGSeasy images are fairly heavy (2–4GB). As a result, we flattened all images in order to compress multiple Docker layers into one, creating an image with fewer and smaller layers, before committing and pushing to Docker Hub.
With exception of the content built into the base image, each NGSeasy pipeline component (Table 2) is encapsulated in a separate container. Using separate containers helps to minimize container size, reduce unexpected interactions between components, and maximise the re-usability of containers.
| Component | Short description | Version |
|---|---|---|
| FastQC21 | Quality reports | 0.11.2 |
| Trimmomatic22 | Read trimmer | 0.32 |
| Picardtools23 | NGS tool | 1.128 |
| GATK19 | NGS tool | 3.2-2 |
| BWA24 | Aligner | 0.7.12-r1039 |
| Bowtie225 | Aligner | 2.2.5 |
| Stampy26 | Aligner | 1.0.23 |
| Snap27 | Aligner | 1.0beta.18. |
| Novoalign28 | Aligner | 3.02.11 |
| Glia29 | Re-aligner | 03-2015 |
| FreeBayes30 | Variant caller | 0.9.21-5 |
| Platypus31 | Variant caller | 0.8 |
A typical NGS pipeline for variant calling and discovery involves the following steps, all of which are implemented in the current version of NGSeasy (1.0-r001):
1. Pre-alignment quality control
2. Sequence alignment
3. Raw alignment processing (e.g. local realignment around candidate indel sites and base quality score recalibration)
4. Post-alignment quality control
5. Variant calling
NGSeasy contains all of the basic tools needed for manipulation and quality control of raw FASTQ files (Illumina focused), SAM/BAM manipulation, alignment, SAM/BAM cleaning and first pass variant discovery. The software we provide as part of NGSeasy are summarised in Table 1 and Table 2.
NGSeasy follows many of the current published best practices for next generation DNA sequencing analysis, specifically, we include options to include the Genome Analysis Toolkit (GATK) recommendations for de-duplication (using Picardtools MarkDuplicates), GATK’s base quality score recalibration (BQSR) and GATK’s realignment around indels18–20.
We also include alternatives to GATK’s BQSR and indel realignment tools, specifically, BamUtil’s recab function http://genome.sph.umich.edu/wiki/BamUtil:recab), and for indel realignment, use of glia (https://github.com/ekg/glia). These options are provided for use in commercial and/or clinical laboratories who do not want to use or pay for a GATK licence.
Containerised software is automatically deployed, so we have opted to provide a wide variety of tools, including multiple tools for alignment and variant calling where available.
To keep the NGSeasy pipeline small and portable, input files, indexed reference genomes and generated output should bypass the container’s root file system instead using a host mounted directory or volume (Figure 1).
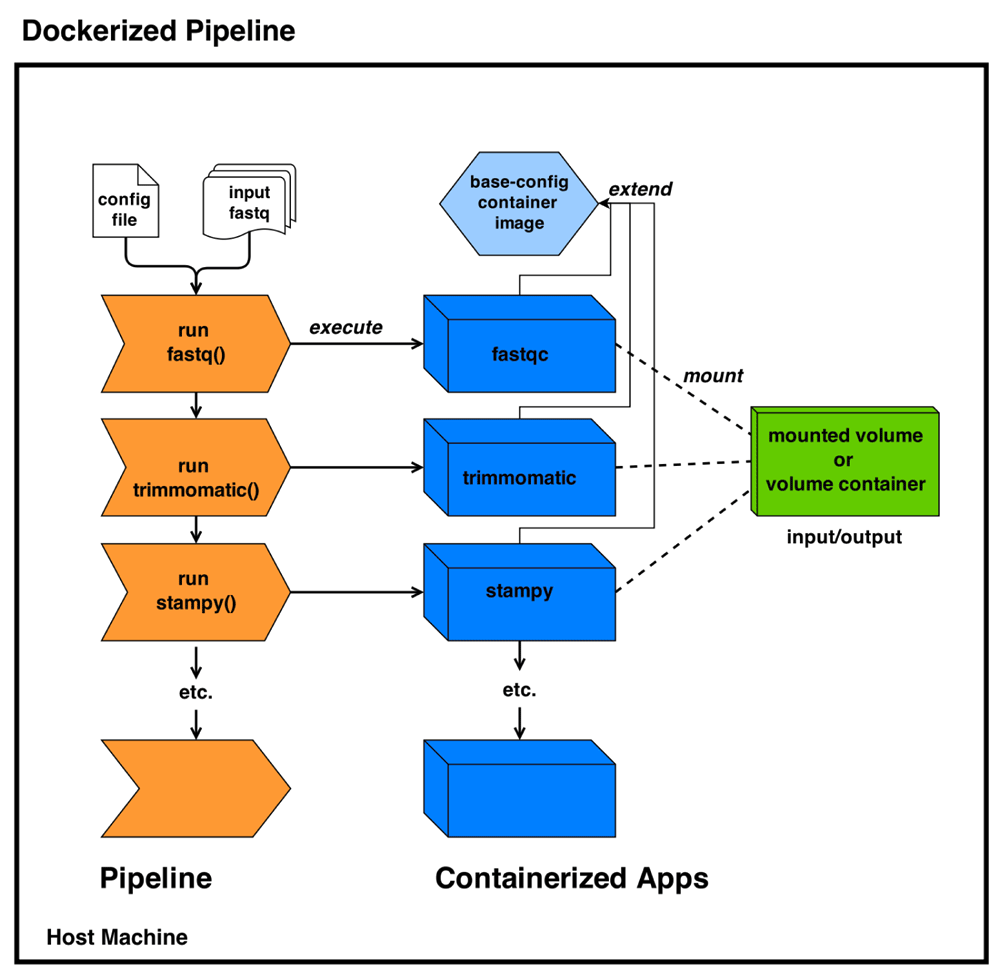
In certain instances it may be necessary to inspect a running container and this can be done by injecting a new process (e.g. a shell terminal) into the container with the docker-exec command, a valuable feature for debugging or monitoring. For resource allocation, Docker uses cgroups to control memory and CPU allocation (hard or soft allocation).
The container images are only provided for software which is freely available. For software components which require registration (e.g. GATK), or are proprietary (e.g. novoalign), we provide a short Dockerfile to complete the build with the additional components which the user must acquire. We believe this is a pragmatic solution for packaging and publishing pipelines that provide the option to use components with a restricted licence. In this way we provide maximum automated deployment with the minimum burden on the end user.
NGSeasy consists of a set of shell (bash) script wrappers, that orchestrate and call all parts of the Dockerised NGS pipeline - where the system calls are to docker run -i -t NGSTool instead of /bin/bash NGSTool, for example. Docker is agnostic, however, in that any workflow management software can be used to orchestrate a Docker based pipeline (eg. rufus2 or nextflow32).
Our design choice was largely influenced by our desire to provide a lightweight and fairly dependency free solution, that is "easy" to set up and maintain. We did not want the user to be tasked with installing a large number of software dependencies before being able to run NGSeasy. In this way, NGSeasy takes advantage of the fact that any modern computer, running any operating system with Docker (or for example boot2docker https://github.com/boot2docker/boot2docker-cli) installed, will come pre-packaged with all of the basic software needed to run a NGS pipeline.
NGSeasy gives the user several options to call a complete NGS pipeline, going from raw FASTQ files to aligned BAM files, variant calls (VCF) and annotations using a range of software. All options are defined in a simple configuration file that can be made, for example, using any spreadsheet application, and then saved as a tab-delimited text file. With this, the user is able to choose from a wide selection of sequence aligners, and variant callers, see Table 2.
The NGSeasy scripts enforce specific naming conventions and directory structures upon the user - allowing sensible and reproducible organisation of NGS projects and associated data on the users local machine. This also avoids all of the potential issues with typographical errors that are typical of manual input.
All NGSeasy applications are run as a non-root user within each container. This is hard coded in the NGSeasy ecosystem and provides some security for Docker containers running in shared computing environments.
Many useful optimisations and recommendations were adapted from bcbio-nextgen (https://bcbio-nextgen.readthedocs.org/en/latest/ - A python toolkit providing best practice pipelines for fully automated high throughput sequencing analysis - and speedseq (https://github.com/cc2qe/speedseq) - a flexible and open source framework to rapidly identify genomic variation33.
For useful cutting edge discussion and testing of NGS pipelines, we also refer readers to the Blue Collar Bioinformatics site at http://bcb.io/.
All Dockerfiles used to generate the NGSeasy images are available at https://github.com/KHPInformatics/ngseasy along with documentation on installing and running NGSeasy. The pre-built containers are available to download from https://registry.hub.docker.com/repos/compbio.
Getting and running NGSeasy is simple and outlined in the code block below.
Listing 1. "Getting and running NGSeasy"
## Install Docker : Full instructions at https://docs.docker.com/ ## Get NGSeasy git clone https://github.com/KHP-Informatics/ngseasy.git ## Install NGSeasy cd ngseasy sudo make INTSALLDIR="/media/scratch" all sudo make intsall ## Running NGSeasy nsgeasy -c ngseasy_test.config.tsv -d /media/scratch/nsg_projects
Users should note that deploying the pipeline containers is fairly fast, dependant on network speeds, however, downloading the reference genomes and test datasets for the resources folder can take a while. For example, the install time averages at about 94 min on machines connected to relatively fast networks (i.e. > 500 Mbit/s).
For full details on obtaining, setting up and running NGSeasy, please refer to our GitHub repository documentation (https://github.com/KHPInformatics/ngseasy).
See Table 3 for our recommended system requirements. The hard disk requirements are based on our experience, and result from the fact that the pipeline/tools produce a range of intermediary and temporary files for each sample. The full NGSeasy install includes indexed genomes for hg19 and b37 for all aligners, annotation files from the GATK’s resource bundle (ftp://ftp.broadinstitute.org/bundle, 34), and all of the NGSeasy Docker images.
Based on our experience, a basic NGS computing system for a small lab would consist of at least 4TB disk space, 60GB RAM and at least 32 CPU cores. Network speed is a major bottle neck when dealing with NGS sized data, and groups are encouraged to think about these issues before embarking on multi sample or population level studies - where computing requirements can very quickly escalate, and transferring NGS data between sites becomes a major rate limiting step.
We tested basic NGSeasy functionality - going from raw .fastq to .bam to .vcf - on an Illumina 100bp paired end whole exome (30x coverage) dataset available from GCAT: Genome Comparison and Analytic Testing - An analytical framework for optimizing variant discovery from personal genomes (http://www.bioplanet.com/gcat). For more details about GCAT, please refer to 35.
For this report, a basic/fast "non-GATK" based pipeline was tested. We skipped FASTQ quality control trimming, re-alignment around indels and BQSR. The selected pipeline first runs FastQC on the raw data, followed by read alignment using all of the selected aligners: stampy, snap, novoalign, and bowtie2. All reads were aligned to the UCSC hg19 reference genome available at http://hgdownload.cse.ucsc.edu/goldenpath/hg19/chromosomes/.
The alignment stage outputs a duplicate marked (samblaster), sorted and indexed BAM file (sambamba), annotated with the appropriate read group information (e.g. sample name, platform unit etc). The alignment stage also includes generation of basic alignment statistics using sambamba’s flagstat function, and a bed file of aligned regions using the bedtools function bamtobed - these extras steps are reflected in the average run times for NGSeasy’s alignment stage (see Table 4). Note that stampy alignment is contingent on aligning reads with bwa first, and hence, we chose not to report separate results for bwa.
Variant calling was peformed using the haplotype based variant callers Platypus31 and FreeBayes30, and the resulting VCF files uploaded to the GCAT server for comparisons to the genome in a bottle (GIB) call set36. The GCAT results for the tests listed above are available at the following urls:
1. All aligners + FreeBayes: http://goo.gl/G9tHRK.
2. All aligners + Platypus: http://goo.gl/CB88G9.
A full discussion on GIB performance statistics is beyond the scope of this paper. Briefly, for the 30x whole exome dataset, NGSeasy is achieving GIB sensitivities and specificities of 81.1–85.8% and 99.996–99.998%, respectively. There are obvious gains to be made by further pipeline optimisations, and the planned inclusion of structural variant callers and variant re-calling and filtering options.
We are presenting these results solely as a "proof of concept". That is, we have successfully Dockerised a full NGS pipeline, that is capable of producing meaningful results, that are comparable with public and "best practice" workflows.
For the testing carried out in this paper, NGSeasy was run on Rosalind, an Openstack private cloud based at Kings College London, using a virtual machine with 256GB RAM and 32 cores. We have also successfully tested NGSeasy on workstations running a wide variety of environments (OSX, Windows 7, Ubuntu 14.04).
Average representative run times for a full NGSeasy pipeline and its components are presented in Table 4.
The obvious winners for alignment, based purely on speed, are bwa and snap. The two software are comparable. The extra run time seen for snap are due to loading/reading of the indexed reference genome. Once this has been done, snap will run at speed, and is the fastest aligner these authors have seen. The reported runtime for stampy is dependent on bwa having been run first.
Note, that fastQC and read quality trimming need only be applied once. After which, the pipeline is set up to test for, and skip these stages, if the have already been run - speeding up subsequent pipeline calls that use the same data. Be aware that run times will vary depending on depth, quality of data, and compute power (e.g. available RAM and CPU).
Both Platypus31 and FreeBayes30, are highly parallelisable and run at speed; Platypus being 6x faster than FreeBayes in our test, but, less sensitive than FreeBayes; the average GIB sensitivity over all aligners from Platypus versus FreeBayes was 82.40% versus 84.15%.
Running a full NGS pipeline using Docker containers had no real noticeable reduction in computing performance (run time) when compared to our original native (non-Dockered) NGS pipeline. The differences are in the milliseconds to seconds range, and largely depend on the underlying system hardware (and data quality). These observations are similar to those reported in 37.
Strikingly, depending on available compute, read depth and the selected pipeline components, the observed runs times indicate that a full clinical NGS pipeline could be run, and achieve actionable results in less than 2 hours. This has major positive implications for molecular diagnostics and projects like the 100,000 Genomes Project (http://www.genomicsengland.co.uk/the-100000-genomes-project/). That is, alignment and variant calling are no longer a major bottle neck. More work is needed to speed up and improve library preparation, sequencing machine run times and solutions for variant annotation, prioritisation and clinical reporting.
NGSeasy demonstrates the utility of Docker as a means to package software used in modular workflows. We envisage NGSeasy as a method for deploying drop-in analyses, in scenarios where data cannot be shared (either for size or privacy reasons) and an analysis must be carried out in-situ. In such cases, using a pipeline like NGSeasy, it is simple to develop an analysis off site, package it and deploy it on computational facilities where access to the data is provided, examples of such scenarios include the 100,000 Genomes Project and Illumina BaseSpace38 Docker ’apps’.
In addition, NGSeasy is being tested across a select group of NHS Labs (under the NHS England Open Source Initiative) for molecular diagnostic and clinical research pipelines. In particular, a version of NGSeasy has been adapted by Viapath at King’s College Hospital (publication pending; personal communication from Dr Barnaby Clark and Dr David Brawand http://www.viapath.co.uk/locations/kings-college-hospital). The advantages being, the ease of use and set up, the built in version control and the ability for audit tracking and reproducibility conferred by the use of Docker and the open source community built around GitHub.
NGSeasy is under continual development. What we demonstrate here is the pre-production release and basic proof of concept evaluation of NGSeasy :a next generation sequencing pipeline in Docker containers. We want to present this to the scientific community at large, especially those working in the bioinformatics domain, and wish to encourage and invite collaboration on NGSeasy and our groups efforts to Dockerise bioinformatic pipelines.
The group is currently working on a GUI for NGSeasy and along with a modular benchmarking suite. In planned extensions, NGSeasy will provide options for consensus calling, trio/family and population based calling pipelines, human leukocyte antigen (HLA) calling, structural variant calling, cancer pipelines, more optimisations, improved logging, and the latest b38 indexed genomes.
In later versions we will publish detailed benchmarking statistics for all aligners and variant calling on whole exome, genome and clinical panels from a range of depths and platforms.
Development work on Docker continues at pace. The present Docker daemon, runs as root, and there remain security issues with the notion of providing access to this daemon in a shared user environment, such as a typical cluster, a solution to this exists using Linux kernel user namespaces but this is presently undergoing review.
1. Container images are available from: https://registry.hub.docker.com/repos/compbio
2. Latest source code, Dockerfiles, pipeline and documentation are available from: https://github.com/KHP-Informatics/ngseasy
3. Link to archived source code as at time of publication: http://dx.doi.org/10.5281/zenodo.3144439
4. GNU General Public License, version 2: http://www.gnu.org/licenses/oldlicenses/gpl-2.0.en.html
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)