Keywords
Neurodegeneration, ubiquitin, proteasome, deubiquitinating enzyme, tau, TDP-43, ubiquitin-specific peptidase 14, protein clearance
This article is included in the Preclinical Reproducibility and Robustness gateway.
Neurodegeneration, ubiquitin, proteasome, deubiquitinating enzyme, tau, TDP-43, ubiquitin-specific peptidase 14, protein clearance
In the new version we have specified the experimental conditions for repeat experiments. Repeated experiments showed similar results, but the conditions in each case were slightly different (i.e., varied concentrations of constructs or a different set of siRNAs were used). Therefore, we were unable to report averages across experiments or perform statistical analyses.
We have updated the discussion to address the comments by Scott Wilson and also the post-review comment from Dr. Kodadek. We cite two publications describing changes in neurodegenerative protein levels (prion protein or huntingtin) after transfection with USP14 or catalytically inactive mutant in vitro. However, it is beyond the scope of this research note to review the literature on IU1.
We changed “arbitrary units” to “tau/actin” or “Flag-TDP-43/actin” as appropriate on the graphs in Figures 1 and 2.
To read any peer review reports and author responses for this article, follow the "read" links in the Open Peer Review table.
Research on the ubiquitin-proteasome system has far reaching implications for the development of drugs to treat illnesses associated with the accumulation of misfolded proteins, including Alzheimer’s and Parkinson’s disease (Ciechanover & Kwon, 2015). Ubiquitin-specific protease 14 (USP14), like its yeast ortholog Ubp6, is a proteasome-associated deubiquitinating enzyme (DUB) that is activated upon binding to the proteasome and catalyzes the cleavage of ubiquitin subunits from substrates before degradation by the proteasome (Borodovsky et al., 2001; Hanna et al., 2006; Hu et al., 2005). By releasing ubiquitin molecules from the substrate, USP14/Ubp6 helps to prevent the rapid degradation of ubiquitin molecules together with the substrate protein (Hanna et al., 2007). A critical role of USP14 in stabilizing cellular ubiquitin levels was demonstrated in vivo in USP14 deficient axJ mice which display decreased ubiquitin levels in all tissues with the greatest loss observed at synaptic terminals (Anderson et al., 2005; Wilson et al., 2002).
In addition to maintaining cellular ubiquitin pools, USP14/Ubp6 has been shown to modulate substrate degradation. Goldberg and colleagues showed that upon binding to a substrate’s polyubiquitin chain, activated USP14/Ubp6 facilitates gate-opening of the proteasome (Peth et al., 2009). This mutual interaction of USP14/Ubp6 with the proteasome is thought to enhance selectivity of the proteasome for ubiquitinated proteins and couple deubiquitination to degradation. In contrast, Finley and colleagues found that USP14/Ubp6, and in some instances a catalytically inactive mutant (C114A in mammals), could cause an inhibition of the degradation of substrates (Hanna et al., 2006; Lee et al., 2010). For model substrates and ataxin3, this effect was shown to require USP14/Ubp6 protein but not its catalytic activity. For two proteins involved in neurodegenerative diseases, tau and TDP-43, inhibition of proteasomal degradation by USP14 was dependent on its deubiquitinating activity, since the catalytically inactive mutant had no effect (Lee et al., 2010). This led to the hypothesis and proof thereof that deubiquitination of substrates by USP14 at a faster rate than the proteasome initiates degradation could cause rejection of otherwise competent substrates from the proteasome (Lee et al., 2016). Inhibition of USP14 by a small molecule inhibitor (IU1) enhanced proteasomal substrate degradation in cells overexpressing tau or TDP-43 (Lee et al., 2010). Thus, inhibition of USP14 was proposed as a therapeutic strategy to enhance proteasomal function in neurodegenerative diseases in which these proteins accumulate.
Constructs. Human USP14 (hUSP14wt), V5-tagged hUSP14wt (V5-hUSP14wt), catalytically inactive mutant USP14-C114A (hUSP14CA), V5-tagged hUSP14CA (V5-hUSP14CA), human tau, and Flag-tagged human TDP- 43 (Flag-TDP-43) were cloned into the pTT5d expression vector by Amgen’s Protein Sciences department and confirmed by sequencing. Human α-synuclein-Flag CMV6 expression vector was purchased from Origene (#RC221446) and confirmed by sequencing.
Cell lines. All cell lines were obtained from ATCC. HEK293 cells were grown in DMEM/10% fetal bovine serum/1% penicillin, streptomycin, glutamine. U2OS cells stably expressing Flag-tagged human α-synuclein (U2OS/synuclein) were generated by Amgen Neuroscience in San Francisco and grown in McCoy’s 5A/10% fetal bovine serum/1% penicillin, streptomycin/2% glutamine and 0.5mg/mL G418. SH-SY5Y cells were grown DMEM/10% fetal bovine serum/1% penicillin, streptomycin, glutamine and 0.5mg/mL G418. All cells were grown in incubators at 5%CO2/37°C. All cell culture reagents were purchased from Gibco.
Transfections. HEK293 cells were plated at a density of 10-6 cells/well in 6-well plates and transfected with plasmids using Lipofectamine™ 2000 (Thermofisher) for 4 hours, and analyzed 48 hours after transfection. U2OS/synuclein cells were plated at 5×10-4 cells/well in 24-well plates and SH-SY5Y cells were plated at 2×10-5 cells/well in 6-well plates. Cells were transfected with Opti-MEM™ (Thermofisher) containing 100nM siRNA, and analyzed 60, 72 or 96 hours after transfection. USP14 siRNAs were obtained from Ambion.
Western blot. Cells were lysed with Lysis Reagent (Roche) containing 1% SDS/1X Complete™ protease inhibitors cocktail tablets (Roche). Samples were boiled and Benzonase Nuclease (Sigma) was added following the manufacturer’s instructions. 10ug of lysate was loaded on a 12% Bis-Tris gel (Life-Sciences) and proteins were separated by electrophoresis (100mA, 200V) and transferred onto 0.2µm nitrocellulose membrane (Life Sciences) for a minimum of 4hrs (100mA, 25V). Membranes were blocked with Odyssey Blocking Buffer (Li-Cor), incubated with primary antibodies diluted in Li-Cor buffer with 0.2% Tween-20 at 4°C shaking overnight, and washed 3× with phosphate-buffered saline/0.1% Tween-20 (PBST). Membranes were then incubated with secondary antibodies for 1 hour at room temperature in the dark, washed 3× with PBST, and analyzed with the Odyssey imaging system at a relative intensity setting of 2–2.5 for the 800 channel and 1–2 for the 700 channel. Beta-actin or GAPDH served as a loading control.
Antibodies. Mouse monoclonal anti-tau5 (1µg/ml; Invitrogen AHB0042), mouse monoclonal beta-actin (1:1000; Cell Signaling 3700S), mouse monoclonal anti-flag (1:500; Sigma-Aldrich F1804), mouse monoclonal anti-V5 (1µg/ml, Sigma-Aldrich V8012), mouse monoclonal anti-GAPDH (1µg/ml; Invitrogen 39–8600), chicken polyclonal anti-USP14 (5µg/ml; Lifesensors AB505), IRDye 680 or 800 anti-mouse or anti-chicken infrared secondary antibodies (1:10000; Li-Cor).
Data analysis. Ratios of the intensity readings for the protein of interest and the loading control were calculated in Microsoft Excel 2010 and plotted using GraphPad Prism 6.05.
A key experiment from Lee et al., 2010, (Figure 1g) showed that recombinantly expressed tau or TDP-43 levels in HEK293 cells were higher when coexpressed with wild type as compared to catalytically inactive (C114A) USP14. We cotransfected V5-pTT5d-USP14 or V5-pTT5d-USP14 (C114A) plasmids (ranging from 0.5 to 2µg) and 2µg pTT5d-Tau or pTT5d-Flag-TDP-43 plasmids in HEK293 cells. Note that we used a pTT5d vector to express proteins, while Finley and colleagues used a pcDNA3.1 vector (Invitrogen). Despite robust expression of USP14 or the catalytically inactive mutant as detected by anti-V5 antibody (Figure 1A, C), no decrease was observed in the levels of tau (Figure 1A, B) or TDP-43 (Figure 1C, D) in cells transfected with the catalytically inactive mutant compared to wild type USP14. A similar experiment was performed in which 1 µg of Tau or TDP-43 was cotransfected with 2 µg USP14 constructs for 48 hours and this also did not appear to alter Tau or TDP-43 levels (not shown).

1, 1.5 or 2ug of V5-hUSP14wt (wt = wild type) or V5-hUSP14(CA) (CA = C114A, catalytically inactive) were cotransfected with 2ug Tau or Flag-TDP-43 plasmid in HEK293 cells. Cells were lysed after 48 hours and analyzed by western blot using a standard protocol. Actin served as loading control. Despite robust expression of USP14 or its catalytically inactive mutant as detected by the V5-tag (A, C), no differences were observed in Tau (A, B) or Flag-TDP-43 (C, D) protein levels. Note that we did not observe differences in the expression levels of USP14 versus USP14(CA). Control = empty vector control.
To exclude the possibility that the V5-tag rendered the USP14 constructs non-functional, we validated an anti-USP14 antibody (Supplementary material) and tested untagged USP14 constructs in TDP-43 overexpressing cells. HEK293 cells were transfected with USP14 or USP14(C114A) plasmids at concentrations ranging from 31ng to 4µg and tau and Flag-TDP-43 at concentrations of 0.5µg; representative blots are shown in Figure 2. Despite robust expression of USP14 or its catalytically inactive mutant as detected by the USP14 antibody, no decrease was observed in tau or Flag-TDP-43 protein levels in cells transfected with the catalytically inactive mutant compared to wild type USP14 (Figure 2A, B). Two similar experiments were conducted with 2 µg Tau or TDP-43 cotransfected with 0.05 or 0.1 µg USP14 constructs or 4 µg Tau or TDP-43 cotransfected with 0.5, 1.0, 2.0 or 4.0 µg of the USP14 constructs; Tau and TDP-43 levels did not appear altered in either experiment (not shown).
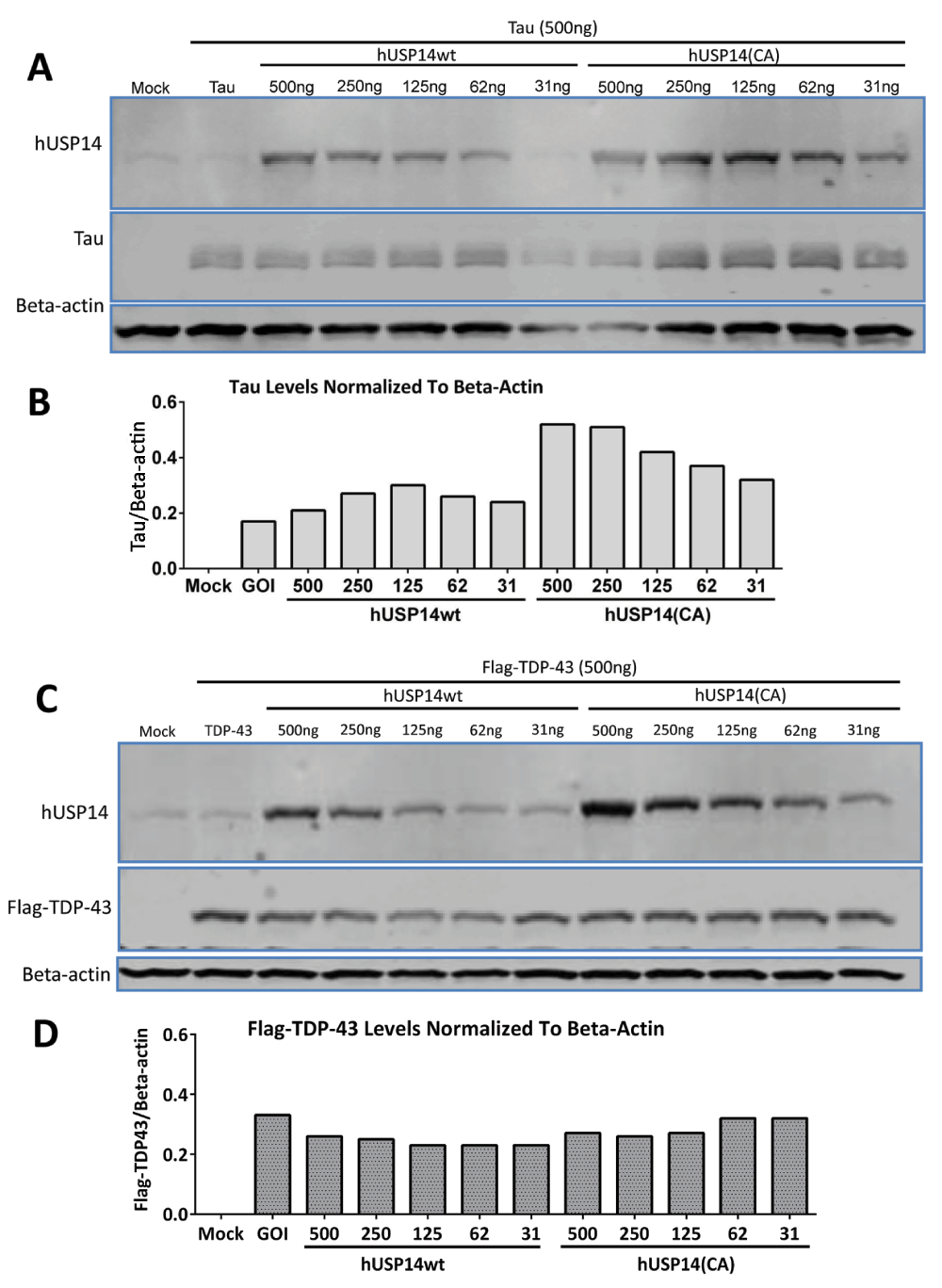
31 to 500ng of hUSP14wt or hUSP14(CA) plasmids were cotransfected with 2ug tau or TDP-43 plasmid in HEK293 cells. Cells were lysed after 48 hours and analyzed by western blot using a standard protocol. Actin served as loading control. Despite robust expression of USP14 or the catalytically inactive mutant as detected by anti-USP14 antibody (A, C), no decreases were observed in tau (A, B) or TDP-43 (C, D) protein levels in the cells transfected with hUSP14CA. Note that we did not observe differences in the expression levels of USP14 versus USP14(CA). Mock = empty vector control, GOI = gene of interest and refers to either tau or TDP-43 in the absence of USP14 cotransfection.
Because there was a possibility that even the untagged-USP14 constructs were not functional, we tested whether siRNA knock down of endogenous USP14 would increase turnover of substrate. Lee et al. (2010) showed that Usp14-/- mouse embryonic fibroblasts had lower levels of tau or TDP-43 than those overexpressing wild-type USP14. Therefore, we reasoned that USP14 knockdown should result in lower levels of substrate. To avoid variability resulting from transient transfections, we tested USP14 knockdown in a stable Flag-tagged α-synuclein U2OS cell line. As shown in Figure 3, four different siRNAs (A58, A59, A60 and A90; 100nM) caused a 50–75% decrease in endogenous USP14 protein levels at 60 or 96 hours post-transfection (Figure 3A, B). No changes in Flag-α-synuclein were detected (Figure 3A, C).
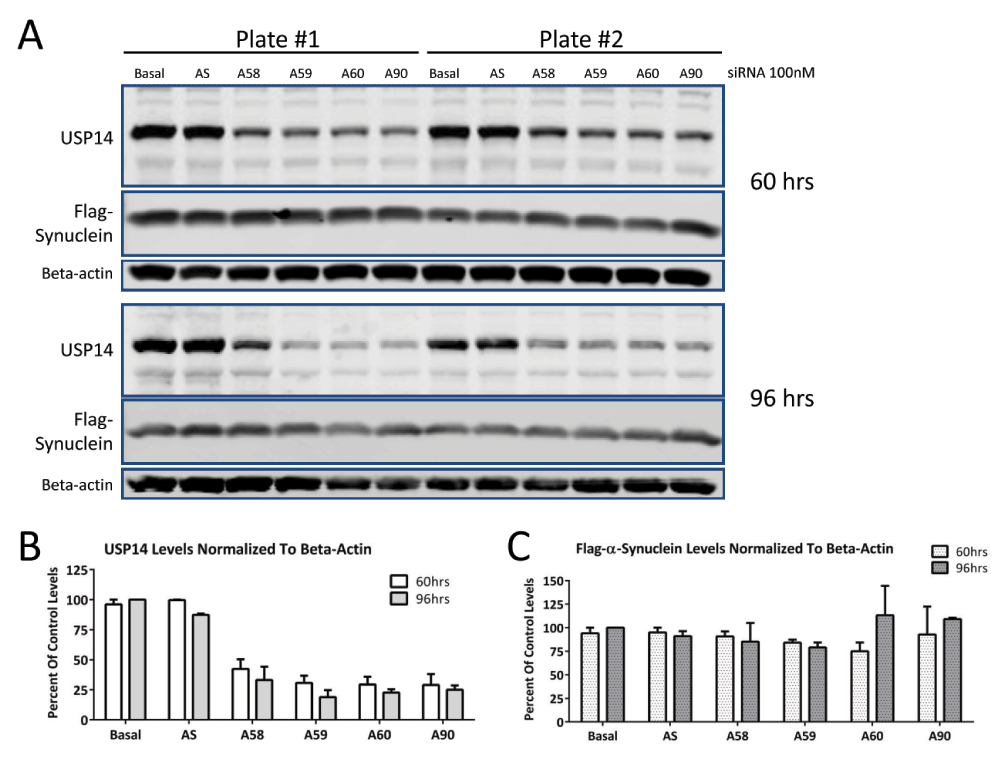
U2OS cells stably expressing Flag-α-synuclein were treated with 100nM USP14 siRNA from Ambion (A58, A59, A60 or A90) for 60 or 96 hours (A). Scrambled siRNA (AS) served as control for the specificity of the siRNA knockdown. Despite 50–75% knockdown of basal USP14 protein levels (B), no changes in Flag-α-synuclein expression were detected (C).
Finally, to eliminate the concern that the artificial levels of the transiently or stably overexpressed substrates caused the lack of effect, we repeated the siRNA knockdown experiment in SH-SY5Y cells that endogenously express tau using siRNAs from Ambion (A58, A59, A60 and A90; 100nM). As shown in a representative western blot in Figure 4, no changes in endogenous tau levels were observed despite a 50–75% knockdown of endogenous USP14 protein levels. This experiment was repeated with four siRNAs from Qiagen at 48 hours and despite a 60–75% knockdown of USP14, we did not observe a consistent relationship between knockdown of USP14 and tau levels (not shown).

SH-SY5Y cells endogenously expressing tau were transfected with 100nM USP14 siRNAs from Ambion (A58, A59, A60 or A90) or scrambled siRNA (AS) for 72 hours. Cells were lysed and analyzed by western blot using a standard protocol. A 50–75% decrease in USP14 protein levels was achieved compared to scrambled control, but no change in basal tau protein levels (A, B).
Though we took several different approaches to assay the effects of USP14 on substrate levels, we were unable to confirm a robust role for USP14 in tau or TDP-43 degradation in our experimental systems. The possibility remains that differences in our methods (such as using a different expression vector) caused the discrepancies between our data and those in Lee et al. (2010). For example, the levels of proteasome-bound USP14 may have differed or protein synthesis and degradation rates may have been altered with our expression system. USP14 might also exert alternative functions that are dependent on substrate or cellular context: In a cellular model of prion disease, overexpression of catalytically inactive USP14 reduced accumulation of prion protein (Homma et al., 2015), whereas in a cellular Huntington’s disease model overexpression of catalytically inactive USP14 had no effect on huntingtin protein aggregates (Hyrskyluoto et al., 2014). Instead, overexpression of wild type USP14 reduced huntingtin aggregation. In USP14-deficient axJ mice in vivo Wilson and colleagues found no changes in endogenous tau or ataxin-3 protein levels, but did observe a difference in phosphorylated tau (Jin et al., 2012). They also generated mice expressing catalytically inactive USP14 and could not detect altered proteasomal function in these mice, although tau levels were not analyzed (Vaden et al., 2015). In combination, these studies highlight the complexity of USP14 biology and future research is needed to unravel the mechanisms that give rise to the apparent discrepancies. We hope our findings serve as a starting point for further discussion, collaboration, and research in this field.
Open Science Framework: Dataset: Does inactivation of USP14 enhance degradation of proteasomal substrates that are associated with neurodegenerative diseases?, doi 10.17605/OSF.IO/7G3MJ (Ortuno et al., 2016).
DO conducted all experiments. DO, HC and SM conceived of the experimental design. HC and SM wrote the article.
All authors were full-time employees at Amgen Inc. at the time the experiments were conducted.
The authors like to thank Amgen’s Protein Sciences department for providing the constructs for transfections and Amgen’s Neuroscience group in San Francisco for providing the stable U2OS/synuclein cell line.
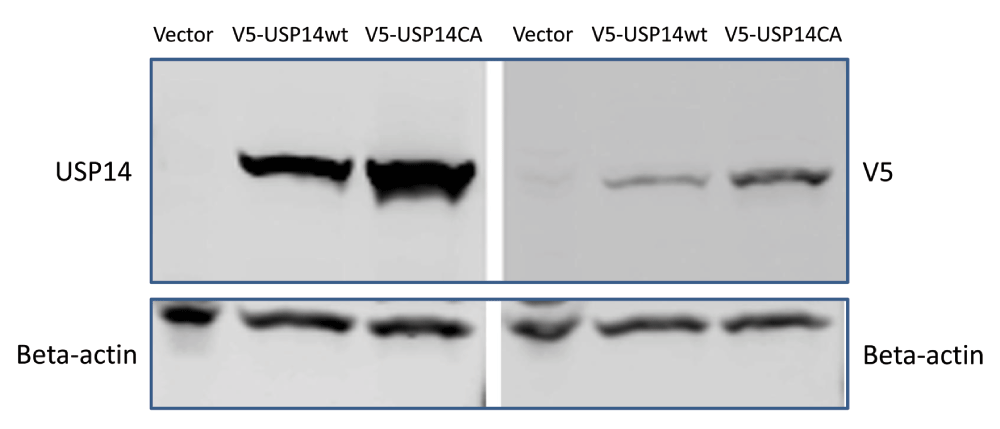
1μg V5-tagged USP14, V5-tagged USP14(CA) or empty vector control constructs were transfected in HEK293 cells and probed with V5 or chicken polyclonal anti-USP14 antibodies. Beta-actin served as loading control.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Most generally, Ortuna et al (2016) pose the question of whether the deubiquitinating enzyme USP14 is in fact a regulator of the proteasome, or if ... Continue reading Response from Daniel Finley:
Most generally, Ortuna et al (2016) pose the question of whether the deubiquitinating enzyme USP14 is in fact a regulator of the proteasome, or if it is, whether the mode of regulation that we uncovered in a series of studies, especially Lee et al Nature 467, 179 (2010), is real. To begin with, here is a list of 38 studies that employ our inhibitors (IU1 or derivatives of IU1) and/or USP14 mutants, revealing accelerated degradation of some protein, as predicted by our model that USP14 and its yeast ortholog Ubp6 are inhibitory to the proteasome (we exclude papers co-authored by any authors of Lee et al 2010):
Jia et al Cell Mol Biol Lett 27:111 [2022]
Hai et al Acta Biochim Biophys Sin Shanghai 54:1 [2022]
Su et al J Biol Chem 299:102734 [2022]
Shi et al Nat Comm 13:5644 [2022]
Pan et al Cell Biocem Biophys doi: 10.1007/s12013-022-01107-y [2022]
Zhao et al Cell Death Differ doi: 10.1038/s41418-022-01040-w [2022]
Wu et al Cell Biol Toxicol doi: 10.1007/s10565-022-09729-x [2022]
Zhu et al Hereditas 159:21 [2022]
Li et al Pharmacol Res 174:105933 [2021]
Xu etal FASEB J 35:e21870 [2021]
Lv et al Cell Death Dis 12:803 [2021]
Yan et al J Biosci 46:19 [2021]
Xu et al Int J Biol Sci 16:2951 [2021]
Shen et al BBRC 524:683 [2020]
Rathore et al Scientific Reports 10:5350 [2020]
Geng et al BBRC 524:16 [2020]
Liu et al PNAS 116:21732 [2019]
Massa et al Hum Mol Genet 28, 764 [2019]
Schattling et al Nature Neurosci 22:887 [2019]
Xia et al J Exp Clin Cancer Res 38:220 [2019]
Liao et al Oncogene 37, 1896 [2018]
Liu et al Nat Comm 9, 4770 [2018]
Chen et al Nature Communications 9, 1223 [2018]
Chakraborty et al EMBO Mol Med 10:e9014 [2018]
VerPlank et al Glia 66:379 [2018]
Wei et al Sci Signal 10:eaak9660 [2017]
Kim and Goldberg, J Biol Chem 292:9830 [2017]
Wei et al Sci Signal 10: pii: eaak9660 [2017]
Liao et al Cell Death Dis 8, e2585 [2017]
Song et al Physiol Biochem 42, 965 [2017]
Min et al J Neurochem 140, 826 [2017]
Zhu et al Oncotarget 8, 48725 [2017]
McKinnon et al Acta Neuropathol 131:411 [2016]
Sareen-Khanna et al Am J Physiol Renal Physiol 311, F1035 [2016]
Chen et al Mol Cell 64, 105 [2016]
Nakashima et al BBRC 472, 598 [2016]
Bashore et al Nat Struct Mol Biol 22:712 [2015]
Homma et al Sci Rep 5:11028. doi: 10.1038/srep11028 [2015]
Doeppner et al Acta Neuropathol 126:251 [2013]
Wu et al Int J Mol Sci 14:10749 [2013]
There is obviously an abundance of observations that are in agreement with the model of Lee et al (2010). Our extensive prior work adds to this body of findings (Hanna et al Cell 127:99 [2006]; Hanna et al Cell 129:747 [2007], and others).
USP14 inhibitors are highly effective in a wide variety of physiological and cellular and disease-related contexts, as discussed below. In support of the paradigm of Lee et al (2010), a remarkable study reported that the severe phenotypes of Parkin and Pink1 mutants in the fly were significantly and specifically suppressed by either loss of function mutations in the gene encoding Usp14 or by simple oral administration of IU1, with dramatic effects on both motor function and longevity (Chakraborty et al EMBO Mol Med 10:e9014 [2018]). The underlying nature of the effect seems to be conserved to mammals (Chakraborty 2018). Another interesting example involving neurodegeneration is myelin protein zero. Deletion of codon 63 from the gene for this protein gives raise to Charcot Marie Tooth 1B disease. In mouse models of this disease, IU1 enhances the rate of degradation of the mutant protein and of overall protein degradation ex vivo in sciatic nerves (VerPlank et al Glia 66:379 [2018]), in accord with the paradigm of Lee et al 2010. A third neurodegenerative disease model, bassoon proteinopathy, a form of multiple sclerosis, was ameliorated phenotypically by IU1 administration in mouse models, and enhanced clearance of the mutant Bassoon protein was observed (Schattling et al Nature Neurosci 22:887 [2019]). Thus, contrary to Ortuna et al (2016), USP14 inhibitors have performed well in multiple models of neurodegenerative disease, including in vivo models in multiple organisms. Importantly, the three examples discussed above provide the most interesting disease models at this point.
The main point of Lee et al (2010) was that specific small-molecule inhibitors of a deubiquitinating enzyme can be identified (against the prevailing view at the time; IU1 was the first specific inhibitor of a deubiquitinating enzyme), and that such inhibitors can be used to accelerate the degradation of specific proteins. It was an important advance for the field and had good generality: it inspired similar work on many other deubiquitinating enzymes. The basis for the specificity of these compounds was elegantly described by Wang et al Cell Research 28:1186 (2018). After extensive medicinal chemistry efforts, USP14 inhibitors of higher potency and selectivity now exist and will hopefully be in the public domain soon.
If contacted, I would have recommended that the Ortuno group try their experiments with our reagents, since it is possible that their various vectors produce levels of expression that obscure the effects. To argue that a study cannot be replicated, it is best to try to replicate the original experimental design. For loss of function studies, Ortuna et al relied on a very weak siRNA knockdown, which is poor practice, since, for many enzymes, phenotypic effects require drastic reduction of their level or activity. There was no indication that the modest knockdown achieved by Ortuna et al was effective functionally, and the data cannot be interpreted with any confidence. I would have advised a more reliable approach, as we used in Lee et al (2010): null mutants. Comments to this effect were subsequently posted on the website of the Ortuno study by Dr. Peter Walter, and elaborated on more fully at https://www.ascb.org/publications-columns/presidents-column/on-reproducibility-and-clocks/.
Interestingly, the USP14 inhibitors reported in Lee et al (2010) have also been found to be cytoprotective in many settings (Sareen-Khanna et al Am J Physiol Renal Physiol 311, F1035 [2016]; Min et al J Neurochem 140, 826 [2017]; Chen et al Nature Communications 9, 1223 [2018]; VerPlank et al Glia 66:379 [2018]; Pan et al Cell Biochem Biophys doi: 10.1007/s12013-022-01107-y [2022]; Schattling et al Nature Neurosci 22:887 [2019]). These studies involve multiple neurodegenerative models, as noted above, neuronal injury via ischemia, and stressed renal cells. Also, consistent with the model of Lee et al (2010), elevated USP14 levels were found to give rise to marked increases in both protein aggregates and amyloid in HCT116 cells (Chen et al Nature Communications 9, 1223 [2018]), consistent with Schattling et al Nature Neurosci 22:887 [2019].
Ortuna et al (2016) entertained a hypothesis contrary to the model of Lee et al (2010), namely that USP14 activates the proteasome instead of inhibiting it. This has since been put to the test by others through examining USP14 knockout MEFs; overall protein degradation was accelerated in the mutant cells, exactly as predicted by our paradigm that USP14 is inhibitory to the proteasome (Kim and Goldberg, J Biol Chem 292:9830 [2017]). This is consistent with the 37 other studies listed above.
It is not the case that, as Ortuna et al argue, the effect on TDP-43 degradation cannot be replicated. See Rathore et al, Scientific Reports 10:5350 [2020], where strong effects were observed both genetically and through the use of IU1 in several human cell lines. This is not to deny that TDP-43 may be a "mediocre substrate" in other contexts (for example if it is not ubiquitinated). Several other reports have confirmed effects on tau levels in various experimental systems, although likewise the strength of the effect may depend on the cell line or on other factors such as the highly heterogeneous nature of tau post-translational modifications (Boselli et al J Biol Chem 292:19209 [2017]; Lee et al Sci Rep 5:10757 [2015]; Kim et al Cell Rep 24:732 [2018]; Singh et al Cell Chem Biol 27: 292 [2020]; Yan et al Cell 185, 3913 [2022]). In general, outcomes can vary depending on the nature of the cells tested, their physiological state, the mutant being tested, the level of expression of target proteins, whether mice or humans and being studied, whether animals of culture cells are being studied, and whether a hypomorph or nullimorph is being studied. Such differences have to do with biology and not with reproducibility. When one group finds such an effect in one context while a group working in a different context does not, it should not in itself be cause for alarmism or be taken to imply that a true contradiction exists.
For the degradation of a protein to be affected by USP14 inhibition, it must not only be a substrate of USP14, it must also be ubiquitinated in the particular cellular, physiological, or organismal context of the experiment. Also, for a significant effect on protein turnover to be seen, Ubp6/USP14 would have to be the dominant or co-dominant deubiquitinating enzyme for the substrate in question. This is likely a major requirement, since there are approximately 100 deubiquitinating enzymes encoded in the human genome. To cite one recent example, a broad screen implicated three deubiquitinating enzymes in the control of tau levels: Usp11, Usp13, and Usp14 (Yan et al Cell 185:3913 [2022]). Such functional redundancy among deubiquitinating enzymes may pertain even where the underlying selectivity mechanisms differ. An effect seen in one cell type might be masked in another because of differences in the activity levels of other deubiquitinating enzymes (not to mention ubiquitin ligases). Inhibitors of deubiquitinating enzymes might often be effective through synergy, even when they seem to score negative when tested individually.
To our best current understanding, USP14 and Ubp6 have a very distinct and novel substrate specificity–they essentially act only on proteins that are ubiquitinated on multiple sites, as we have shown (Lee et al Nature 532:398 [2016]). Even a protein carrying a long ubiquitin chain is not detectably deubiquitinated by USP14/Ubp6. This may explain why many proteasome substrates are not USP14 substrates. But when a substrate is properly modified at multiple sites, USP14/Ubp6 can remove chains en bloc at a rapid rate, measurable on a millisecond time scale, as we have shown, until only a single chain remains (Lee et al Nature 2016). This explains how USP14/Ubp6 can suppress degradation by the proteasome–it is capable of acting significantly faster than the proteasome, even for good proteasome substrates. But USP14/Ubp6 (especially Ubp6) also inhibits degradation in a noncatalytic mode (Hanna et al Cell 127:99 2006; Hanna et al Cell 129:747 [2007]), by inducing the proteasome to adopt unique conformational states, and this is now understood in detail through cryo-EM studies of both USP14-proteasome and Ubp6-proteasome co-complexes (Zhang et al Nature 605:567 [2022]; Hung et al Nature Comm 13:838 [2022]). In their discussion, Ortuna et al seem to argue that altered proteasome function is difficult or impossible to detect in USP14 mutants, contrary to abundant evidence in the literature. We have observed these effects repeatedly for over 15 years; other laboratories have reported the same effects (Bashore et al Nat Struct Mol Biol 22:712 [2015]--also predating Ortuna et al). In summary, extensive understanding of USP14/Ubp6 has been achieved, including strong experimental support that these enzymes have the capacity to inhibit proteasome function and protein degradation as we have proposed (Lee et al 2010).
Most generally, Ortuna et al (2016) pose the question of whether the deubiquitinating enzyme USP14 is in fact a regulator of the proteasome, or if it is, whether the mode of regulation that we uncovered in a series of studies, especially Lee et al Nature 467, 179 (2010), is real. To begin with, here is a list of 38 studies that employ our inhibitors (IU1 or derivatives of IU1) and/or USP14 mutants, revealing accelerated degradation of some protein, as predicted by our model that USP14 and its yeast ortholog Ubp6 are inhibitory to the proteasome (we exclude papers co-authored by any authors of Lee et al 2010):
Jia et al Cell Mol Biol Lett 27:111 [2022]
Hai et al Acta Biochim Biophys Sin Shanghai 54:1 [2022]
Su et al J Biol Chem 299:102734 [2022]
Shi et al Nat Comm 13:5644 [2022]
Pan et al Cell Biocem Biophys doi: 10.1007/s12013-022-01107-y [2022]
Zhao et al Cell Death Differ doi: 10.1038/s41418-022-01040-w [2022]
Wu et al Cell Biol Toxicol doi: 10.1007/s10565-022-09729-x [2022]
Zhu et al Hereditas 159:21 [2022]
Li et al Pharmacol Res 174:105933 [2021]
Xu etal FASEB J 35:e21870 [2021]
Lv et al Cell Death Dis 12:803 [2021]
Yan et al J Biosci 46:19 [2021]
Xu et al Int J Biol Sci 16:2951 [2021]
Shen et al BBRC 524:683 [2020]
Rathore et al Scientific Reports 10:5350 [2020]
Geng et al BBRC 524:16 [2020]
Liu et al PNAS 116:21732 [2019]
Massa et al Hum Mol Genet 28, 764 [2019]
Schattling et al Nature Neurosci 22:887 [2019]
Xia et al J Exp Clin Cancer Res 38:220 [2019]
Liao et al Oncogene 37, 1896 [2018]
Liu et al Nat Comm 9, 4770 [2018]
Chen et al Nature Communications 9, 1223 [2018]
Chakraborty et al EMBO Mol Med 10:e9014 [2018]
VerPlank et al Glia 66:379 [2018]
Wei et al Sci Signal 10:eaak9660 [2017]
Kim and Goldberg, J Biol Chem 292:9830 [2017]
Wei et al Sci Signal 10: pii: eaak9660 [2017]
Liao et al Cell Death Dis 8, e2585 [2017]
Song et al Physiol Biochem 42, 965 [2017]
Min et al J Neurochem 140, 826 [2017]
Zhu et al Oncotarget 8, 48725 [2017]
McKinnon et al Acta Neuropathol 131:411 [2016]
Sareen-Khanna et al Am J Physiol Renal Physiol 311, F1035 [2016]
Chen et al Mol Cell 64, 105 [2016]
Nakashima et al BBRC 472, 598 [2016]
Bashore et al Nat Struct Mol Biol 22:712 [2015]
Homma et al Sci Rep 5:11028. doi: 10.1038/srep11028 [2015]
Doeppner et al Acta Neuropathol 126:251 [2013]
Wu et al Int J Mol Sci 14:10749 [2013]
There is obviously an abundance of observations that are in agreement with the model of Lee et al (2010). Our extensive prior work adds to this body of findings (Hanna et al Cell 127:99 [2006]; Hanna et al Cell 129:747 [2007], and others).
USP14 inhibitors are highly effective in a wide variety of physiological and cellular and disease-related contexts, as discussed below. In support of the paradigm of Lee et al (2010), a remarkable study reported that the severe phenotypes of Parkin and Pink1 mutants in the fly were significantly and specifically suppressed by either loss of function mutations in the gene encoding Usp14 or by simple oral administration of IU1, with dramatic effects on both motor function and longevity (Chakraborty et al EMBO Mol Med 10:e9014 [2018]). The underlying nature of the effect seems to be conserved to mammals (Chakraborty 2018). Another interesting example involving neurodegeneration is myelin protein zero. Deletion of codon 63 from the gene for this protein gives raise to Charcot Marie Tooth 1B disease. In mouse models of this disease, IU1 enhances the rate of degradation of the mutant protein and of overall protein degradation ex vivo in sciatic nerves (VerPlank et al Glia 66:379 [2018]), in accord with the paradigm of Lee et al 2010. A third neurodegenerative disease model, bassoon proteinopathy, a form of multiple sclerosis, was ameliorated phenotypically by IU1 administration in mouse models, and enhanced clearance of the mutant Bassoon protein was observed (Schattling et al Nature Neurosci 22:887 [2019]). Thus, contrary to Ortuna et al (2016), USP14 inhibitors have performed well in multiple models of neurodegenerative disease, including in vivo models in multiple organisms. Importantly, the three examples discussed above provide the most interesting disease models at this point.
The main point of Lee et al (2010) was that specific small-molecule inhibitors of a deubiquitinating enzyme can be identified (against the prevailing view at the time; IU1 was the first specific inhibitor of a deubiquitinating enzyme), and that such inhibitors can be used to accelerate the degradation of specific proteins. It was an important advance for the field and had good generality: it inspired similar work on many other deubiquitinating enzymes. The basis for the specificity of these compounds was elegantly described by Wang et al Cell Research 28:1186 (2018). After extensive medicinal chemistry efforts, USP14 inhibitors of higher potency and selectivity now exist and will hopefully be in the public domain soon.
If contacted, I would have recommended that the Ortuno group try their experiments with our reagents, since it is possible that their various vectors produce levels of expression that obscure the effects. To argue that a study cannot be replicated, it is best to try to replicate the original experimental design. For loss of function studies, Ortuna et al relied on a very weak siRNA knockdown, which is poor practice, since, for many enzymes, phenotypic effects require drastic reduction of their level or activity. There was no indication that the modest knockdown achieved by Ortuna et al was effective functionally, and the data cannot be interpreted with any confidence. I would have advised a more reliable approach, as we used in Lee et al (2010): null mutants. Comments to this effect were subsequently posted on the website of the Ortuno study by Dr. Peter Walter, and elaborated on more fully at https://www.ascb.org/publications-columns/presidents-column/on-reproducibility-and-clocks/.
Interestingly, the USP14 inhibitors reported in Lee et al (2010) have also been found to be cytoprotective in many settings (Sareen-Khanna et al Am J Physiol Renal Physiol 311, F1035 [2016]; Min et al J Neurochem 140, 826 [2017]; Chen et al Nature Communications 9, 1223 [2018]; VerPlank et al Glia 66:379 [2018]; Pan et al Cell Biochem Biophys doi: 10.1007/s12013-022-01107-y [2022]; Schattling et al Nature Neurosci 22:887 [2019]). These studies involve multiple neurodegenerative models, as noted above, neuronal injury via ischemia, and stressed renal cells. Also, consistent with the model of Lee et al (2010), elevated USP14 levels were found to give rise to marked increases in both protein aggregates and amyloid in HCT116 cells (Chen et al Nature Communications 9, 1223 [2018]), consistent with Schattling et al Nature Neurosci 22:887 [2019].
Ortuna et al (2016) entertained a hypothesis contrary to the model of Lee et al (2010), namely that USP14 activates the proteasome instead of inhibiting it. This has since been put to the test by others through examining USP14 knockout MEFs; overall protein degradation was accelerated in the mutant cells, exactly as predicted by our paradigm that USP14 is inhibitory to the proteasome (Kim and Goldberg, J Biol Chem 292:9830 [2017]). This is consistent with the 37 other studies listed above.
It is not the case that, as Ortuna et al argue, the effect on TDP-43 degradation cannot be replicated. See Rathore et al, Scientific Reports 10:5350 [2020], where strong effects were observed both genetically and through the use of IU1 in several human cell lines. This is not to deny that TDP-43 may be a "mediocre substrate" in other contexts (for example if it is not ubiquitinated). Several other reports have confirmed effects on tau levels in various experimental systems, although likewise the strength of the effect may depend on the cell line or on other factors such as the highly heterogeneous nature of tau post-translational modifications (Boselli et al J Biol Chem 292:19209 [2017]; Lee et al Sci Rep 5:10757 [2015]; Kim et al Cell Rep 24:732 [2018]; Singh et al Cell Chem Biol 27: 292 [2020]; Yan et al Cell 185, 3913 [2022]). In general, outcomes can vary depending on the nature of the cells tested, their physiological state, the mutant being tested, the level of expression of target proteins, whether mice or humans and being studied, whether animals of culture cells are being studied, and whether a hypomorph or nullimorph is being studied. Such differences have to do with biology and not with reproducibility. When one group finds such an effect in one context while a group working in a different context does not, it should not in itself be cause for alarmism or be taken to imply that a true contradiction exists.
For the degradation of a protein to be affected by USP14 inhibition, it must not only be a substrate of USP14, it must also be ubiquitinated in the particular cellular, physiological, or organismal context of the experiment. Also, for a significant effect on protein turnover to be seen, Ubp6/USP14 would have to be the dominant or co-dominant deubiquitinating enzyme for the substrate in question. This is likely a major requirement, since there are approximately 100 deubiquitinating enzymes encoded in the human genome. To cite one recent example, a broad screen implicated three deubiquitinating enzymes in the control of tau levels: Usp11, Usp13, and Usp14 (Yan et al Cell 185:3913 [2022]). Such functional redundancy among deubiquitinating enzymes may pertain even where the underlying selectivity mechanisms differ. An effect seen in one cell type might be masked in another because of differences in the activity levels of other deubiquitinating enzymes (not to mention ubiquitin ligases). Inhibitors of deubiquitinating enzymes might often be effective through synergy, even when they seem to score negative when tested individually.
To our best current understanding, USP14 and Ubp6 have a very distinct and novel substrate specificity–they essentially act only on proteins that are ubiquitinated on multiple sites, as we have shown (Lee et al Nature 532:398 [2016]). Even a protein carrying a long ubiquitin chain is not detectably deubiquitinated by USP14/Ubp6. This may explain why many proteasome substrates are not USP14 substrates. But when a substrate is properly modified at multiple sites, USP14/Ubp6 can remove chains en bloc at a rapid rate, measurable on a millisecond time scale, as we have shown, until only a single chain remains (Lee et al Nature 2016). This explains how USP14/Ubp6 can suppress degradation by the proteasome–it is capable of acting significantly faster than the proteasome, even for good proteasome substrates. But USP14/Ubp6 (especially Ubp6) also inhibits degradation in a noncatalytic mode (Hanna et al Cell 127:99 2006; Hanna et al Cell 129:747 [2007]), by inducing the proteasome to adopt unique conformational states, and this is now understood in detail through cryo-EM studies of both USP14-proteasome and Ubp6-proteasome co-complexes (Zhang et al Nature 605:567 [2022]; Hung et al Nature Comm 13:838 [2022]). In their discussion, Ortuna et al seem to argue that altered proteasome function is difficult or impossible to detect in USP14 mutants, contrary to abundant evidence in the literature. We have observed these effects repeatedly for over 15 years; other laboratories have reported the same effects (Bashore et al Nat Struct Mol Biol 22:712 [2015]--also predating Ortuna et al). In summary, extensive understanding of USP14/Ubp6 has been achieved, including strong experimental support that these enzymes have the capacity to inhibit proteasome function and protein degradation as we have proposed (Lee et al 2010).