Keywords
Neuromuscular disease, neurodegeneration, ALS, FTD, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, Primary Lateral Sclerosis
Neuromuscular disease, neurodegeneration, ALS, FTD, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, Primary Lateral Sclerosis
The author wishes to thank the referees for their valuable comments. These have been included in version 2 of the manuscript as follows:
To read any peer review reports and author responses for this article, follow the "read" links in the Open Peer Review table.
The pathogenesis of most neuromuscular and neurological diseases is poorly understood, despite their devastating impact on quality of life and the fact that they were first described more than a century ago. Clinically, neuromuscular diseases manifest as progressive muscle weakness together with a general set of motor symptoms, including speech-related difficulties, impaired mobility, and reduced fine motor skills1. In contrast, neurological diseases manifest primarily as a progressive decline in cognitive function. Interestingly, the clinical manifestations of neuromuscular and neurological diseases also overlap; this overlap is summarized in Table 1 for primary lateral sclerosis (PLS), amyotrophic lateral sclerosis (ALS), ALS with frontotemporal dementia (ALS-FTD), FTD with ALS (FTD-ALS), FTD, Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease1–29. The clinical features shared between the neuromuscular disease ALS and the neurological disease FTD exemplify this overlap, as late-stage ALS can lead to the manifestation of FTD; conversely, FTD can progress to ALS, leading to the manifestation of FTD-ALS2–8.
A key observation gleaned from analyzing Table 1 is the finding that glutamate levels are increased in the cerebrospinal fluid (CSF) of patients in all eight diseases22–29. Glutamatergic (i.e., excitatory) overstimulation induces excitotoxicity in cultured neurons and is believed to be an important factor in the pathogenesis of both neuromuscular and neurological diseases22–29. Glutamate-induced excitotoxicity can result in the decay of neuronal pathways that innervate muscles and other physiological systems22–29. This decay gives rise to the loss of physiological function and is considered to lead to the clinical manifestations that present with both neuromuscular disease and neurological disease22–29. I hypothesize that these increased glutamate levels are actually a homeostatic response to an overstimulated inhibitory nervous system. This novel hypothesis is based upon the observation that the clinical findings in neuromuscular and neurological diseases can be explained by inhibitory activity, as discussed below.
Since they were first diagnosed more than a century ago, the clinical manifestations of neuromuscular and neurologic diseases have been well described. Strikingly, however, the consequences of one key clinical feature of these diseases—the absence of an elevated risk of seizure activity—have been largely overlooked.
This is exemplified for ALS in which a broad, detailed retrospective study of the medical records of 657 ALS patients revealed that none of the patients presented with epilepsy as a co-morbid condition9. Moreover, a thorough search of PubMed for articles published from 1966 through 2016 using the key words “seizure” or “epilepsy” in combination with “amyotrophic lateral sclerosis” or “ALS” confirms the striking absence of epilepsy and/or seizures in ALS patients. This finding is consistent with the absence of seizures and/or epilepsy in review articles describing the clinical manifestation of ALS2–6.
A key observation that makes the absence of seizure activity in ALS even more remarkable is increased glutamate levels in the cerebrospinal fluid (CSF) of patients with ALS; on average, glutamate levels in the CSF of ALS patients are increased by 100%, and some ALS patients can have an increase of up to 800%23. Importantly, increased glutamate levels are generally associated with epileptic seizures30,31. Thus, given the increased glutamate levels typically measured in the CSF of ALS patients, one would logically expect that the prevalence of epilepsy in ALS patients should be elevated relative to the general population. However, despite this expectation, epileptic seizures are simply not reported among ALS patients.
Strikingly, in addition to ALS, none of the other seven diseases listed in Table 1 typically present with an increased risk of epileptic seizures, either2–15, even though all eight diseases present with elevated glutamate levels in the CSF22–29. With respect to Alzheimer’s disease, patients in the early stages of the disease occasionally develop seizures14,32; however, this seizure activity decreases as the disease progresses from the early stages to more advanced stages14,32,33.
A second key observation is that neuromuscular and neurological diseases have an inhibitory effect on muscle function, rather than being excitatory. The diseases listed in Table 1 are characterized by muscle inhibition, even though glutamate—which, as discussed above, is generally increased in these diseases—is the major neurotransmitter that drives muscle activation by increasing the firing rate of motor neurons. Remarkably, however, despite having increased levels of glutamate in the CSF, patients with neuromuscular and neurological diseases do not have increased muscle activation. This is exemplified most clearly by ALS, a disease with highly elevated glutamate levels22,23 and complete muscle inhibition in the end stages. Although fasciculation and/or cramps can be observed in ALS patients2–4, these features occur in debilitated muscles as they progress from a fully functional state toward a fully inhibited state.
One possible explanation for these seemingly contradictory findings is a second system that exerts strong anticonvulsive activity in both neuromuscular and neurological diseases. Importantly, such a system should be as widespread throughout the nervous system as the glutamatergic system, and the inhibitory system fulfills these requirements. Specifically, the inhibitory (GABA) system i) functions to oppose the glutamatergic excitatory neurotransmitter system, ii) inhibits muscle activity by reducing the firing rate of motor neurons, and iii) exerts strong anticonvulsive activity26,30,31.
A third key observation is that the clinical manifestations of neuromuscular and neurological diseases can be induced using interventions that increase GABAergic (i.e., inhibitory) activity (Table 2). For example, activating the GABAergic inhibitory system using benzodiazepines can render healthy muscles dysfunctional34,35. In addition, fatal respiratory depression can be induced by administering an overdose of the GABAergic benzodiazepine midazolam36. Chronically stimulating the inhibitory system can cause chronic muscle disuse that can lead to muscle atrophy37. Moreover, ingestion of alcohol (another GABAergic inhibitory compound38) impedes coordination and causes slurred speech (dysarthria), which are features of neuromuscular and neurological diseases. In cats, dysphagia (difficulty swallowing) can be either induced or reversed using GABA agonists or GABA antagonists, respectively39. Dysphagia has also been reported in humans following the administration of either benzodiazepines40–42 or alcohol43. Administration of benzodiazepines reduces voluntary saccadic eye movement function44 and increases EEG beta-wave activity44, clinical manifestations that also occur in neuromuscular and neurological diseases18,45,46. Increased GABAergic inhibitory activity can also cause bladder47,48 and gastrointestinal dysfunction49,50, both of which can manifest in neuromuscular and neurological diseases19–21. Interestingly, Bravo et al. reported that chronically feeding mice the lactic acid bacterium L. rhamnosus increases expression of GABA receptors, suggesting a link between GABAergic activity and CNS disorders51. Strikingly, GABAergic activity can also explain the overlapping clinical manifestations between Alzheimer’s disease and alcohol-related dementia52, and it can explain the increase in dementia-like symptoms observed after the administration of the benzodiazepine diazepam53. Inhibitory activity can also explain neuromuscular and neurological disease predisposition in the elderly, as the sensitivity to GABA inhibitory activity is known to increase with age54. Finally, GABAergic activity has been implicated in cognitive dysfunction55–57, which is a hallmark feature of neurological diseases and is often observed in late-stage neuromuscular disease2–7. Taken together, these findings support the notion that the clinical features associated with neuromuscular and neurological diseases can be induced by activating the inhibitory system.
In addition to mimicking the majority of clinical manifestations observed in neuromuscular and neurological diseases, GABAergic activity can also explain the more rapid disease progression of ALS reported in clinical trials in which patients received GABAergic compounds. For example, in two trials gabapentin increased the rate of disease progression in patients with ALS58. A similar effect was reported in patients with ALS who received the GABAergic compound topiramate59. GABAergic action can also explain the more rapid disease progression of ALS in clinical trials in which patients received the antibiotic minocycline60, which has GABAergic activity61. Finally, GABAergic involvement can explain the observed efficacy of the taurine conjugate form of ursodeoxycholic acid (UDCA) in ALS patients62, as UDCA inhibits GABAergic action63.
A fourth key observation is that the clinical manifestations associated with neuromuscular and neurological diseases can be attributed to the activity of either simple inhibition (SI) or recurrent inhibition (RI) pathways. Specifically, I postulate that differences between muscles under the control of SI and/or RI underlie the important—yet poorly understood—manifestations of neuromuscular and neurological diseases.
The inhibitory system functions via both SI and RI64. The RI system controls physiological functions that play a role in counteracting gravitational forces and other external forces acting on the body. During locomotion and/or to counteract the effects of gravity, RI uses a negative inhibitory feedback loop (Figure 1), thereby providing muscles with additional, stabilizing input. Therefore, muscles involved in movement and lifting heavy objects are subject to RI. Examples of RI-innervated muscles include the limb and thorax muscles, as well as the neck muscles that control head movement.
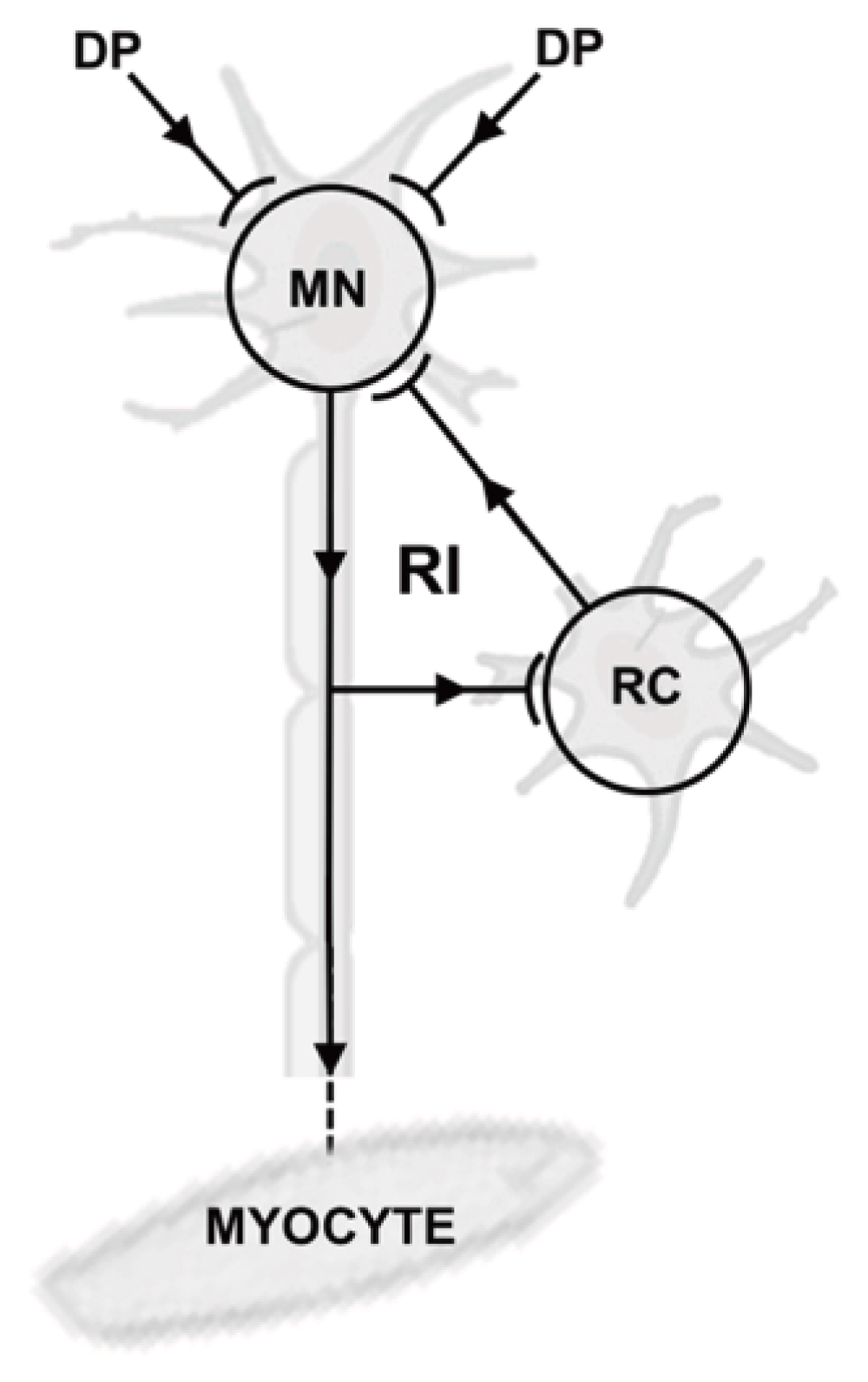
With recurrent inhibition (RI), input from descending pathways (DP) reaches the motor neuron (MN). In response, the MN activates the target myocyte; in addition, the MN also activates Renshaw cells (RC), which then inhibit the motor neuron through a negative feedback loop.
In contrast, neuronal pathways that do not play a role in locomotion or counteracting gravity selectively utilize SI64. Examples of SI-innervated muscles include facial, speech, pharyngeal, and eye muscles, as well as muscles that are involved in bowel and bladder function.
Table 3 summarizes the involvement of SI and RI in the principal clinical manifestations of neuromuscular and/or neurological diseases. A close examination of Table 3 reveals that one set of muscles—namely, the respiratory muscles—is innervated by both SI and RI pathways64,65. This dual innervation arises because the respiratory muscles play a role in both respiratory function and maintaining body posture64.
Strikingly, the categorization between SI-innervated and RI-innervated muscles coincides with the categorization of muscles affected in limb-onset ALS, bulbar-onset ALS, and respiratory-onset ALS. Approximately 70%, 25%, and 5% of ALS patients present initially with limb involvement (limb-onset ALS), bulbar symptoms (bulbar-onset ALS), or respiratory symptoms (respiratory-onset ALS), respectively2, and this difference in onset can be explained by differences in SI versus RI involvement (Table 4). Patients with both SI and RI involvement at the onset of disease present with respiratory-onset ALS. In contrast, patients with primarily SI involvement present with bulbar-onset ALS, whereas patients with primarily RI involvement present with limb-onset ALS (see Table 4). Thus, SI and RI differentiation can account for this difference in ALS onset.
The differential involvement of the SI or RI system can also explain the observed differences in life expectancy between patients who present with limb-onset, bulbar-onset, or respiratory-onset ALS. Specifically, patients with respiratory-onset ALS generally have the shortest life expectancy following diagnosis4. As discussed above, increased activity of both the SI and RI pathways leads to fatal respiratory depression, the principal cause of early death in patients with ALS.
Increased activity of either the SI or RI pathway—but not both—can also lead to respiratory depression, albeit not to fatal levels. Under these conditions, respiratory function, though impaired, can be maintained by either SI or RI pathway activity. However, due to impaired respiratory function resulting from either SI or RI overstimulation (see Table 4), these patients can die from dysphagia-related malnutrition and/or aspiration pneumonia. Thus, patients with either SI or RI involvement—but not both—generally live longer than patients with both SI and RI involvement. Importantly, this observation can also explain why patients with motor neuron diseases at either end of the SI/RI spectrum—for example, primary lateral sclerosis, progressive muscular atrophy, progressive bulbar palsy, or pseudobulbar palsy—have a longer life expectancy than patients with ALS4, which lies in the middle of the spectrum.
Differences between the effects of SI versus RI involvement can explain the fact that although ALS and FTD generally involve two distinct systems, these two diseases have a certain degree of overlap with respect to their clinical manifestations (see Table 1). Thus, if RI overstimulation precedes SI involvement, the patient can present with an initial diagnosis of ALS and can progress to ALS-FTD, a common manifestation of cognitive dysfunction observed in 20–50% of patients with late-state ALS patients2–7. Alternatively, if SI overstimulation precedes RI involvement, FTD is the initial diagnosis, and the disease can progress to FTD-ALS when the RI pathway becomes involved. Moreover, the division between SI and RI can also explain the overlap between subcategories of ALS and FTD with respect to impaired cognition and altered behavior that involve SI, and movement dysfunction that involves RI.
Although it is generally considered one disease, ALS can present with a wide spectrum of clinical manifestations, and this spectrum can be explained by the involvement of SI and/or RI pathways. For example, SI overstimulation can lead to bulbar, cognitive, and frontotemporal dementia-related manifestations without causing severe muscle wasting or respiratory malfunction (for example, as observed in patients with bulbar-onset ALS). On the other hand, RI overstimulation can lead to locked-in syndrome, a state in which the patient retains cognitive and emotional function but becomes “locked” in their body, with all of the muscles that counteract gravity and other external forces rendered essentially dysfunctional. Interestingly, the only muscles that are spared in locked-in syndrome—and the only way in which end-stage patients can communicate with the outside world—are the muscles that control eye movement. This is an important observation, given that the muscles that control eye movement are not controlled by RI pathways (see Table 3). The distinction between SI and RI can also explain the observation that some patients with ALS have fully intact cognitive and emotional functions even after their muscles involved in countering gravity have become dysfunctional; the most famous example of this phenomenon is Stephen Hawking, who despite being diagnosed with ALS in his early twenties remains active as a prominent theoretical physicist, now in his seventies.
Split-hand syndrome is common among patients with ALS66. With split-hand syndrome, the abductor pollicis brevis (APB) and first dorsal interosseous (FDI) muscles are affected, whereas the abductor digiti minimi (ADM) muscle is relatively spared. This syndrome is particularly puzzling, as these muscles are innervated identically, yet are affected differently66. I propose that split-hand syndrome can be attributed to differences in the extent to which RI pathways innervate the hand muscles that are involved in precision gripping, versus muscles that also play a role in power gripping. With precision gripping (for example, when using a pen), the fingers and thumb press against each other; this type of grip does not involve lifting a relatively heavy object67. In contrast, power gripping (for example, when gripping a hammer or lifting a heavy pan) uses the fingers, palm, and thumb to clamp down on a heavy object in order to lift and control the object67. Napier67 used this distinction to distinguish muscle activities that are involved in body locomotion and/or posture from muscle activities that do not involve locomotion or posture. Thus, Napier’s separation also categorizes muscle activities into those that are controlled by RI and those that are controlled by SI. Because the primary function of the ADM muscle is to move the little finger (i.e., the fifth digit) away from the hand, the ADM muscle is only involved in precision gripping and would therefore not be affected by RI overstimulation. This is consistent with the reported absence of RI in motor neurons that innervate the ADM64,68. On the other hand, the APB and FDI muscles are involved in the opposition and extension of the thumb and are therefore involved in power gripping64; thus, these two muscles are affected by RI overstimulation.
In Parkinson’s disease, rest tremors arise from involuntary rhythmic oscillatory movements of a body part at rest; these tremors stop when the patient actively moves the affected body part. The pathways that underlie rest tremors have not been identified, and the fact that rest tremors resolve during voluntary movement is one of the most puzzling observations associated with Parkinson’s disease69. However, because these tremors occur at rest (and not during active motion or while countering the effects of gravity), the muscles involved are likely innervated by SI pathways, making rest tremors an SI-specific phenomenon. This is further illustrated by the finding that rest tremors resolve when the affected body part becomes involved in locomotion, stance, or maintaining inertia69, actions that involve muscles that are controlled by RI64. Interestingly, the hand tremor that is most specific to patients with Parkinson’s disease—the so-called “pill-rolling tremor”—also results from muscles that are innervated solely by SI pathways. Specifically, the pill-rolling tremor involves muscles that play a role in precision gripping but not in power gripping, a distinction that is highly reminiscent of split-hand syndrome in ALS (see above). Furthermore, involvement of the inhibitory system in Parkinson’s disease rest tremors is supported by the observation that the rest tremors observed in restless legs syndrome resolve after the administration of quinine (FDA Drug safety communication, 2010), a compound that reduces inhibitory activity70.
The differentiation between SI and RI summarized in Table 3 can explain the three categories of fatal symptoms that arise in end-stage neuromuscular and neurological diseases (Table 5). One striking observation from Table 5 is that both SI and RI can be attributed to fatal respiratory failure, the major cause of death among ALS patients. Overstimulation of SI pathways leads to bowel dysfunction, bladder dysfunction, and dysphagia-related malnutrition and aspiration pneumonia; these symptoms are the major causes of death among patients with FTD, Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease. On the other hand, overstimulation of RI pathways can lead to end-stage locked-in syndrome.
Differential SI and RI involvement can also account for the wide variety of clinical manifestations in neuromuscular and neurological diseases during disease progression. As a group, neuromuscular and neurological diseases present with a wide spectrum of clinical manifestations (see Table 1), and stimulation of SI and/or RI pathways can account for this spectrum. For example, SI overstimulation can lead to FTD, Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease, whereas RI overstimulation can lead to locked-in syndrome. Finally, overstimulation of both the SI and RI pathways can lead to ALS.
Finally, the differentiation between SI and RI can help explain the differences in life expectancy among patients with various neuromuscular and neurological diseases. As discussed above, increased activity of both the SI and RI pathways leads to fatal respiratory depression (see Table 5), the principal cause of death in patients with ALS, the neuromuscular disease with the shortest life expectancy. Increased activity of either the SI or RI pathway—but not both—can also lead to respiratory depression, albeit not to direct fatal levels. Thus, patients with either SI or RI overstimulation generally live longer than patients with both SI and RI overstimulation. This coincides with the observation that patients with ALS—in which both the SI and RI pathways are overstimulated—have a shorter life expectancy than patients with FTD, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and locked-in syndrome, diseases in which either SI or RI activity is increased.
Taken together, the wealth of observations discussed above suggest that the opposing excitatory and inhibitory systems may play a role in the pathogenesis of the same disease. This phenomenon has precedent, as inhibitory/excitatory homeostasis processes are also involved in seizure activity30,31. Neurons that receive excessive excitatory stimulation can subsequently become overstimulated by inhibitory transmission, and vice versa. This raises the intriguing question of which system in neuromuscular and neurological diseases is overstimulated first, and which system becomes overstimulated as a homeostatic response. This question has been addressed with respect to epileptic seizures30. With respect to neuromuscular and neurological diseases, it is important to note that the administration of glutamatergic excitatory compounds does not lead to the clinical manifestations summarized in Table 1; glutamatergic overstimulation can give rise to clinical manifestations only through excitotoxicity (i.e., overstimulation-induced neuronal cell death). However, inhibitory overstimulation can give rise to the clinical manifestations in Table 1, even in the absence of neuronal cell death. Thus, I hypothesize that inhibitory overstimulation occurs first, and excitatory overstimulation is a homeostatic response. As inhibitory overstimulation increases, the excitatory system is stimulated further, until it reaches a level that induces neuronal cell death. This process is depicted schematically in Figure 2. Importantly, the order of the homeostatic process hypothesized here is precisely opposite to the homeostatic processes observed during epileptic seizures, in which excitatory overstimulation proceeds inhibitory overstimulation30.

In the inhibitory overstimulation hypothesis, excitatory overstimulation is a homeostatic response to inhibitory overstimulation. A key feature of this model is that inhibitory overstimulation can be sufficient to cause symptoms (left blue arrow). As the disease progresses, increasing inhibitory overstimulation can eventually lead to excitatory overstimulation and neuronal cell death, making the symptoms irreversible.
Other possible interpretations of the findings summarized in Table 1 should be considered. If inhibitory overstimulation plays a key role in neuromuscular and neurological diseases, one would expect these patients to present with sedation, thus indicating the possibility that physiological systems other than the inhibitory system may be involved. However, not all GABA receptor subtypes are involved in sedation71,72. Thus, inhibitory activity can occur without inducing pronounced sedation. This is supported by reports that benzodiazepine-induced dysphagia can occur even in non-sedated patients40–42. Second, the absence of seizures despite high glutamate levels could be due to a slow, but non-epileptogenic increase in glutamate levels during the progression of neuromuscular and neurological diseases. However, even small increases in glutamate levels can increase glutamatergic synchronization of a small subset of critical neurons, thereby leading to epileptic activity30,31. Moreover, epileptic seizures are simply not reported among ALS patients, an observation that cannot be explained by a slow increase in glutamate levels, as glutamate does not cause inhibitory activity22–29. Furthermore, slow increasing levels of glutamate in the absence of seizure activity may reflect the involvement of an inhibitory homeostatic process30,31. Finally, the differentiation between SI and RI depicted in Table 4 and Table 5 may be attributed to the involvement of neuronal pathways projecting to either voluntary or involuntary muscles. However, this cannot explain split-hand syndrome in ALS patients or rest tremors in Parkinson’s disease, as these phenomena involve only voluntary muscles. Moreover, split-hand syndrome involves identically innervated muscles that cannot be differentiated by any aspect other than SI/RI innervation.
From a clinical perspective, an important consequence that emerges from the inhibitory overstimulation hypothesis is that the clinical manifestations summarized in Table 1 develop before neurons have undergone cell death. The implication of this possibility is that decreasing inhibitory activity may be beneficial in terms of slowing—or even preventing—the progression of neuromuscular and neurological diseases. Compounds that can reduce inhibitory activity are currently available; unfortunately, however, these compounds can induce seizure activity and are therefore not used therapeutically. Nevertheless, their potential for preventing the pathogenesis of neuromuscular and neurological diseases suggests that compounds that target the inhibitory system could be developed for clinical applications. For example, the average life expectancy of a patient with ALS is 3–5 years after onset, and most neuromuscular and neurological diseases are severe and ultimately fatal. With respect to Alzheimer’s disease and Parkinson’s disease, dysphagia and respiratory depression–related aspiration pneumonia are the most common causes of death16,17. Neuromuscular diseases also present with the severe and potential fatal clinical manifestations listed in Table 1;1–7,9–11,13,15,17–21 thus, the ability to prevent these symptoms could significantly prolong the life expectancy of these patients. From a treatment perspective, it is interesting to note that the selective GABA antagonist SGS-742 has been shown to be both clinically feasible and safe55.
Based upon the plethora of observations regarding the inhibitory system, I hypothesize that this system plays an important role in the pathogenesis of both neuromuscular and neurological diseases. Importantly, overstimulation of the inhibitory system can explain both the absence of epileptic seizures despite the elevated glutamate levels and the pharmacological induction of symptoms present in patients with neuromuscular and neurological diseases. Moreover, the separation between SI and RI can account for the various categories of clinical manifestations observed in these patients. Specifically, I hypothesize that increased glutamate levels in neuromuscular and neurological diseases are actually a homeostatic response to an overstimulated inhibitory system. Implicating the inhibitory system in the pathogenesis of neuromuscular and neurological diseases is highly relevant, given that the majority of approaches being developed for treating these diseases focus on reducing glutamatergic activity, rather than reducing inhibitory activity. Moreover, this putative connection between the inhibitory system and neuromuscular/neurological diseases may have long-reaching implications, including the need to develop therapies designed to reduce inhibitory overstimulation in neuromuscular and neurological patients.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)