Keywords
Heparan sulphate, heparanase, exosomes, Cell surface proteoglycans,
Heparan sulphate, heparanase, exosomes, Cell surface proteoglycans,
Proteoglycans are present in all cellular and tissue compartments. Moreover, in mammals they are expressed by virtually all cells. By definition, proteoglycans consist of a core protein to which one or more glycosaminoglycan chains are covalently attached. While the number of proteoglycan core proteins in the mammalian genome is not large, their form and functions are highly variable. Aggrecan, a major constituent of cartilage matrix, for example, may have >100 chondroitin sulphate chains, which are key to its function in the maintenance of a hydrated, compression-resisting matrix1,2. Decorin, on the other hand, with roles in collagen fibril formation and regulation of innate immunity, has only one chondroitin or dermatan sulphate chain3. Not surprisingly, since proteoglycans can be intracellular, cell surface, or extracellular matrix components, they are increasingly studied in the context of tumour growth, the tumour and stem cell niche, and invasion, metastasis, and tumour-host interactions4–9.
On the surfaces of most mammalian cells are representatives of two major families of heparan sulphate proteoglycans (HSPGs), the glypicans and syndecans5,10–12. The former are linked to the membrane through a glycosylphosphatidylinositol anchor, while the syndecans are transmembrane, with a highly conserved short cytoplasmic domain. Usually the core proteins carry two to five heparan sulphate chains, but syndecans may sometimes also, or alternately, carry chondroitin or dermatan sulphate chains5. The synthesis of heparan sulphate chains is a complex Golgi apparatus-localised process; while all of the transferases and other modifying enzymes involved in their synthesis are known, their regulation is not13. The importance of heparan sulphate synthesis lies in the fact that this glycosaminoglycan has an ability to interact with a wide array of binding partners that include cytokines, chemokines, growth factors, extracellular matrix macromolecules, enzymes, and lipoproteins14,15. Heparan sulphate chains have regions of high modification (i.e. high levels of sulphation) interspersed with regions of low, or no, sulphation15. This most complex of all post-translational modifications is under scrutiny, since most protein binding partners of heparan sulphate engage with highly sulphated domains14,16, so the control of its synthesis and how this may change with transformation are important issues. Moreover, mature heparan sulphate chains can be further modified by a single mammalian heparanase enzyme and by two sulphatases that selectively remove the sulphates of some glucosamine residues17–19. Heparan sulphate editing is now a topic of great interest in tumour biology and some recent developments are summarised below.
For many years, it was assumed that cell surface HSPGs had few independent functions but were mostly acting in cis as co-receptors with other receptors, e.g., tyrosine kinase growth factor receptors and integrins5,11,12,20. The notion was that the heparan sulphate chains provided binding sites for ligands that could then be concentrated for high-affinity receptor binding and subsequent signalling. It now seems clear that there are more intricate interactions at the cell surface that involve independent roles for the cell surface HSPGs. Some of the latest insights into cell surface HSPG functions with relevance to tumour biology are briefly reviewed here. Recent information on the roles of other classes of extracellular matrix proteoglycans in cancer can be found elsewhere3,4,7,9,21.
There is abundant evidence that heparan sulphates, owing to their diversity in structure and location, play important roles in regulating the growth and progression of cancer. Much of this regulation occurs via the ability of heparan sulphate to fine-tune molecular interactions that regulate cell behaviour22. Over the last decade, it has become increasingly apparent that enzymes can edit heparan sulphate structure, thereby precisely modulating its function and regulating cell behaviour. These enzymes include the endoglucuronidase heparanase, which cleaves and shortens heparan sulphate chains of proteoglycans that as a consequence possess new non-reducing termini, and the extracellular sulphatases Sulf-1 and -2 that selectively remove 6-O sulphates. Both of these enzyme activities are proving to be powerful regulators of tumour behaviour.
Heparanase is associated with aggressive tumour behaviour including enhanced growth, angiogenesis, and metastasis. Although a number of studies in many tumour types have supported these conclusions, a unifying mechanistic explanation of precisely how heparanase promotes angiogenesis and metastasis was lacking until recently. In a paper just published in Oncogenesis, Jung et al. demonstrate that heparanase-mediated trimming of syndecan-1 heparan sulphate chains and upregulation of matrix metalloproteinase-9 (MMP-9) expression results in enhanced shedding of syndecan-1 from the cell surface. Shedding exposes a juxtamembrane site on the syndecan-1 core protein that binds to both very late antigen-4 (VLA-4 [integrin α4β1]) and vascular endothelial growth factor receptor-2 (VEGFR2). This coupling of VLA-4 to VEGFR2 activates the latter, thereby initiating downstream signalling that displaces the cytoskeletal adaptor protein paxillin from VLA-4, in turn facilitating the activation of Rac GTPase and polarised cell migration23. This mechanism is in play on both endothelial cells and tumour cells and demonstrates how heparanase, in concert with syndecan-1, drives angiogenesis, tumour cell invasion, and subsequent metastasis.
Evidence is also emerging that heparanase plays a key role in promoting chemoresistance. In breast cancer cell lines expressing a high level of heparanase, inhibition of the enzyme sensitised the cells to killing by lapatinib24. Elevated heparanase expression by myeloma cells enhances their resistance to both bortezomib and melphalan and this resistance is reversed in vivo when mice are treated with the heparanase inhibitor Roneparstat25. Furthermore, heparanase was shown to be present at a high level on tumour cells that survive extensive chemotherapy in myeloma patients, lending further support to the notion that heparanase promotes resistance to therapy25. Together, these findings raise the exciting possibility that the efficacy of anti-cancer drugs may be enhanced when combined with the use of heparanase inhibitors. This is of particular interest, as there are currently four anti-heparanase drugs in clinical trials in cancer patients19. These drugs are all heparin mimetics that are thought to inhibit heparanase activity by blocking the enzyme’s active site. However, recent solving of the crystal structure of heparanase provides an opportunity for the discovery of small molecule inhibitors of enzyme activity that should exhibit improved specificity over the heparin mimetics26. Heparanase-neutralising antibodies have also recently shown promise in attenuating the growth and metastasis of lymphoma and myeloma tumours in mice27.
While heparanase may have important roles in supporting tumour angiogenesis, it is important to recognise that it is not the only mechanism. Many angiogenesis-promoting growth factors, such as VEGF, fibroblast growth factors (FGFs), cytokines, and chemokines, have high affinity for heparan sulphate. It is therefore likely that vascular remodelling is a consequence of multiple interactions involving cell surface HSPGs14,28–30.
Although it is generally agreed that the function of Sulf-1 and -2 is to selectively remove 6-O sulphates from heparan sulphate chains, the impact of these two extracellular sulphatases on tumour growth and progression remains controversial. By altering the composition of heparan sulphates, the Sulfs regulate the signalling capacity of heparin-binding growth factors such as Wnts, FGF, EGF, and VEGF, among others19. Predictably, this has important consequences for tumour behaviour. What is surprising is that despite their seemingly identical function, there are data to support the conclusion that Sulf-1 suppresses tumour growth while Sulf-2 promotes tumour growth31,32. However, such a generalisation appears to be misleading because there is evidence that in some instances Sulf-1 promotes, while Sulf-2 inhibits, tumour growth. Together, these findings strongly suggest that there are factors beyond the catalytic activity of the Sulfs that determine their ultimate impact on tumour behaviour31,33,34 (Figure 1). Such factors may be related to spatial or temporal expression of the Sulfs, variations in their specificity for the heparan sulphate substrate, or differing abilities of the Sulfs to diffuse through the tumour microenvironment. Moreover, there is evidence for non-catalytic properties of Sulfs that lead to alterations in heparan sulphate synthesis through changes in sulphotransferase expression33 or upregulation of glypican-3 core protein, which is relevant to hepatocellular carcinoma34.
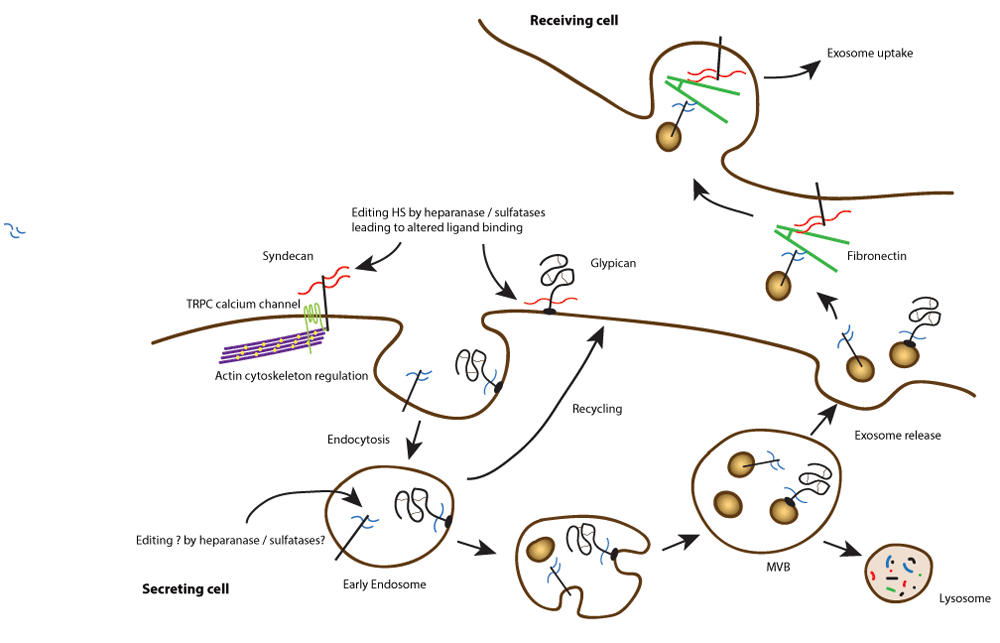
Cell surface heparan sulphate proteoglycans can interact with multiple ligands through their glycosaminoglycan chains. In addition, they can be modified by heparanase and sulphatases, leading to altered ligand binding. Endocytosis, trafficking, and processing can lead to the release of exosomes bearing modified proteoglycans. These can interact with fibronectin in the extracellular environment and ultimately be bound and internalised by recipient cells. This signalling at a distance may be important in the regulation of tumour cell behaviour.
In 2012, the first of several papers was published suggesting that syndecans were cell surface receptors important in exosome formation35. For this, the most C-terminal region of the syndecan cytoplasmic domain interacting with PDZ domain proteins was required. The cytoplasmic scaffolding protein syntenin (also known as melanoma differentiation-associated gene 9; MDA-9) binds to all syndecans through one of its two PDZ domains36,37, and this was shown to be important for the endosomal and trafficking events that lead to exosome formation38. The other PDZ domain of syntenin had high affinity for the membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PtdIns4,5P2). Syntenin also interacts through its C-terminal domain with Bro1/ALG-2-interacting protein (ALIX39), a central player in exosome formation. In turn, ALIX links to a multiprotein endosomal sorting complex required for transport (ESCRT), with additional roles for the GTPase Arf6 and phospholipase D240. Exosomes are now recognised as important signalling vesicles, containing a number of proteins, lipids, and even nucleic acids such as RNAs and miRNAs. They are produced by most cells, including tumour cells, and interest in them from the tumour perspective focuses on whether they can be detectable biomarkers in fluids and their potential roles in regulating the tumour environment (Figure 1). Moreover, syntenin (MDA-9) was first identified in the context of melanoma but is upregulated in many tumours where experiments have shown that it supports cell migration or invasion37,41. It has many binding partners beyond syndecans, including the tetraspanin CD63, an exosome marker42, but what controls the selectivity of syntenin to interact with many different cell surface molecules is currently unclear. However, it has been suggested that this protein is a potential tumour target43.
Interestingly, similar to their roles in regulating tumour angiogenesis and metastasis, heparanase and syndecans also work together in regulating exosome secretion by tumour cells. Enhanced heparanase expression in tumour cells stimulates exosome biogenesis, alters exosome protein composition, and enhances the ability of exosomes to promote tumour cell spreading and endothelial cell migration44. In this instance, heparan sulphate chains of syndecans are essential for exosome formation within endosomal compartments, and trimming of heparan sulphate by heparanase activates the formation of an endosomal complex containing syndecan coupled to syntenin and ALIX35,45. This complex promotes endosomal membrane budding and drives exosome biogenesis. Following their secretion, exosomes exert their biological activity by docking with recipient cells and delivering cargo that can alter recipient cell behaviour. In this context, the heparan sulphate present on syndecan, which remains on the exosome surface following the biogenesis process, can interact with fibronectin via its Hep-II heparin-binding domain46. The fibronectin-coated exosomes subsequently dock by binding to the heparan sulphate of proteoglycans present on the recipient cell surface. At least in some cases, the heparan sulphate present on recipient cells can also act as an internalising receptor, thus facilitating the uptake of exosomes and subsequent delivery of exosome cargo within the cell47 (Figure 1).
Syndecans are not the only proteoglycans with potential importance to exosomes. In 2015, a very interesting report documented that circulating exosomes containing glypican-1 could potentially identify patients with pancreatic ductal adenocarcinoma, even at early stages of tumour development48. Whether the heparan sulphate chains were present and carrying important growth factors, cytokines, or chemokines remains speculative, but once more the connection between cell surface HSPGs and cancer is apparent.
The four mammalian syndecans all interact with the actin cytoskeleton5. Much research has been devoted to understanding this relationship, and many reports have provided evidence that they contribute to microfilament organisation in adhesion and migration. Perhaps the best example in this regard is syndecan-4. It promotes the assembly of focal adhesions, junctions that form in response to cell adhesion to the extracellular matrix. They are integrin-dependent organelles, but the mechanism by which syndecan influences the process has taken many years to unravel. Key to syndecan-4’s role are interactions with both the actin-associated protein α-actinin49–51 and protein kinase Cα, through which there are multiple potential pathways involving Rho family GTPases to the cytoskeleton52,53. The roles of RhoA, Rac, and cdc42 are well known in this regard54,55. Analysis of fibroblasts derived from syndecan-4 null mice show clear differences in microfilament organisation, with much reduced focal adhesions and stress fibres51,56,57, for which RhoGTPase activities seem not to provide the whole explanation. Recent analysis has now shown that this altered adhesion phenotype of S4KO cells relates to calcium channels of the TRPC (transient receptor potential canonical) family. Indeed, elimination of the TRPC7 channel (itself a focal adhesion component) reverts the S4KO cells to wild-type in terms of adhesion, cytoskeleton, and junction formation58. This was accompanied by reductions in cytosolic calcium that were shown to be increased in the null cells compared to matching wild-type cells. Further work with epithelial cells and, moreover, genetic experiments with Caenorhabditis elegans (which possesses a single syndecan) show that this regulation of TRPC type channels by syndecans may be a highly conserved and important role for this proteoglycan family58.
The work with syndecans and channels has so far not embraced tumour cells. Since calcium is a potent regulator of the actin cytoskeleton, it may now be attractive to re-examine some of the previous observations on HSPGs and tumour cells. The literature is replete with studies showing that syndecans are often mis-expressed in solid tumours and in some cases relate to prognosis59–62. A good example is breast cancer, where high levels of syndecan-1 expression, particularly in the tumour stroma, are an indicator of poor prognosis63,64. In other studies, syndecan-2 upregulation has been shown to alter the adhesion and invasiveness of MDA-MB231 breast carcinoma cells and colon carcinoma cells65,66. The difficulty with many studies is understanding whether syndecan expression merely correlates with or is functionally related to tumour progression. In some cases, however, the situation is clearer. A wealth of evidence now suggests that syndecan-1 expression in myeloma is related directly to disease severity and progression67,68. Moreover, it is not only syndecans that may influence tumour progression. Evidence has accumulated rapidly over the past few years showing a relationship between glypican-3 expression and the progression of hepatocellular carcinoma69–71. This HSPG is expressed in foetal liver, but levels subside in postnatal life72,73. However, in a large majority of cases, glypican-3 is re-expressed in hepatocellular carcinoma72,74. The excitement about this HSPG revolves around the possibility that it may serve as a prognostic marker, but also a target for immunotherapy71. Early clinical trials have been reported, but clearly there is a long way to go. On a molecular level, it has been suggested that glypican-3 can bind both Wnt and Frizzled, the signalling receptor for Wnts, through its heparan sulphate chains70,71. However, the situation is complex, since glypican-3 in normal tissue may be a growth inhibitor. Rare core protein mutations giving rise to the Simpson-Golabi-Behmel syndrome are characterised by overgrowth and many dysmorphisms in patients and a corresponding murine model75. In hepatocellular carcinoma, however, there is also upregulation of Sulf-2. It now appears that selective removal of 6-O-sulphate residues from the glypican’s heparan sulphate chains leads to Wnt activation, possibly through its enhanced mobility, leading to Frizzled binding and signalling76. It is also possible that the heparan sulphate chains may bind hepatocyte growth factor and members of the FGF family77,78.
Recent developments have highlighted that both the heparan sulphate chains and the core proteins of cell surface HSPGs are highly and functionally relevant to tumour progression. Moreover, the increasingly recognised importance of the tumour cell niche79,80, which is rich in proteoglycans, and the emerging roles of proteoglycans in stem cell differentiation6,81 are areas for future development. Moreover, it is not only HSPGs that present as targets in tumours. The chondroitin sulphate proteoglycan 4 (also known as NG2) is recognised as a cell surface marker of pericytes in the vasculature but is also present more widely, for example on neuronal and oligodendrocyte precursors82. It is also an emerging target for immunotherapy in a variety of tumour types, including melanoma, triple negative breast cancer, glioblastoma, mesothelioma, and sarcomas83,84.
The potential for cell surface proteoglycans to be targets for intervention are complicated by their multiple roles and ubiquity. It is perhaps likely that tumour cells, stromal/other host tissue, and the immune system utilise these proteoglycans and their downstream signalling in specific ways to regulate behaviour. Targeting will require detailed understanding, and therefore we can predict that new insights into the functions of proteoglycans will impact tumour biology for many years to come.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)