Keywords
supervised learning, complex, Bayesian network, protein-protein interaction, search
This article is included in the Cytoscape gateway.
supervised learning, complex, Bayesian network, protein-protein interaction, search
Protein-protein interaction (PPI) networks provide information about the biochemical relationships in a cell’s molecular machinery. Each node in the network represents a protein, with edges connecting proteins that physically interact with one another. Complexes are clusters of interacting proteins in the network which are together responsible for some biological function. The discovery of complexes in a PPI network has implications in the study of the molecular basis of diseases, drug targets, and biological pathways1.
Existing tools for complex detection in PPI networks rely on assumptions about a common topography observed among most protein complexes. These tools often assume that protein complexes can be identified based on the density of interactions (edges) among the set of its nodes. As a result, the problem is often reduced to clique detection2. While many complexes often take the form of edge-dense protein clusters, this is not the only observed topology. Real-valued data indicate that a variety of other features, including but not limited to edge density, are typical of complexes among PPI networks3.
SCODE, an application for supervised complex detection, seeks to make more informed predictions about potential clusters in PPI networks. It applies supervised learning, a strategy for making predictions based on training data that has already been labeled (as a complex or a non-complex, for example). In order to predict novel complexes, SCODE uses a naive Bayesian classifier to determine the likelihood that potential protein clusters are true complexes.
SCODE allows the user to define a set of features that determine whether a protein cluster forms a complex or not. The probability distribution of this set of features, dependent on whether or not a cluster is a complex, is encoded using naive Bayesian networks. Based on information in the training data, this probability distribution determines which predicted protein clusters conform to the observed features of known complexes. Not only does this identify more qualified candidate complexes, it also provides the user with more autonomy to describe what characteristics are important.
Overall, SCODE performs four major functions: (1) train a Bayesian network, (2) search a PPI graph for candidate complexes, (3) score candidate complexes, and (4) optionally, evaluate the quality of the returned complexes. Figure 1 illustrates the pipeline of these functions in SCODE.
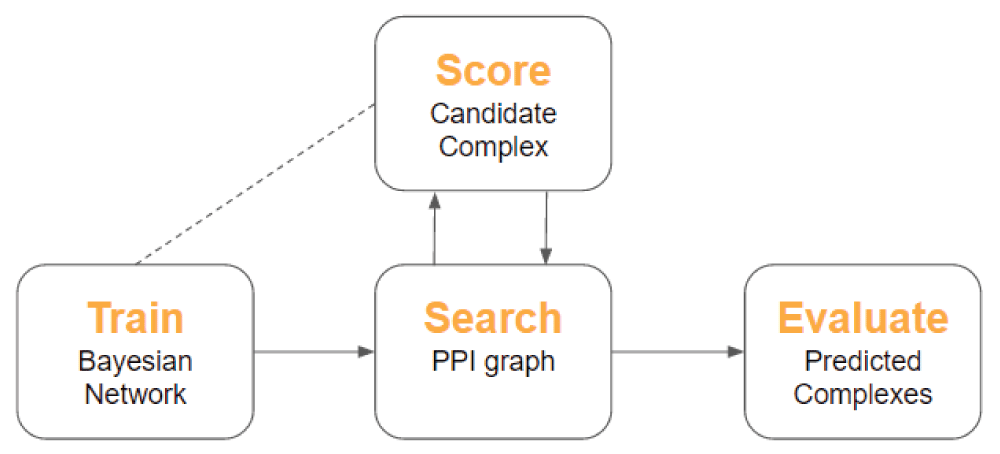
The search algorithm uses a form of iterative simulated annealing in which complexes are expanded to include neighboring proteins at each cycle. The user may specify a set of "seed" proteins from which to begin the search, or allow the program to randomly select a set of starting nodes.
Beginning from each of the seed proteins, the program iteratively performs the following steps:
1. Maintain a record of the neighbor that produces the highest score when added to the complex. For each neighbor of the seed:
2. If the record exceeds the current score of the complex, then add the associated protein to the complex
3. If the record does not exceed the current score of the complex, then calculate the probability of adding the protein to the complex as a function of the score and the temperature. SCODE then adds the protein to the complex with probability as follows: P(exp(Record – Score(complex))/Temperature)
Three variations on this algorithm are available: iterative simulated annealing (ISA), greedy iterative simulated annealing (Greedy ISA), and sorted-neighbor iterative simulated annealing (Sorted-Neighbor ISA). In ISA, at each iteration a node is randomly selected from the neighbors of the candidate complex and scored in order to determine if it should be kept in the complex. Greedy ISA scores all neighbors of the candidate complex and retains only the best one (or none, if no neighbors improve the score). Sorted-Neighbor ISA sorts the neighbors of the complex by degree and evaluates the top N neighbors (N is a parameter set by the user).
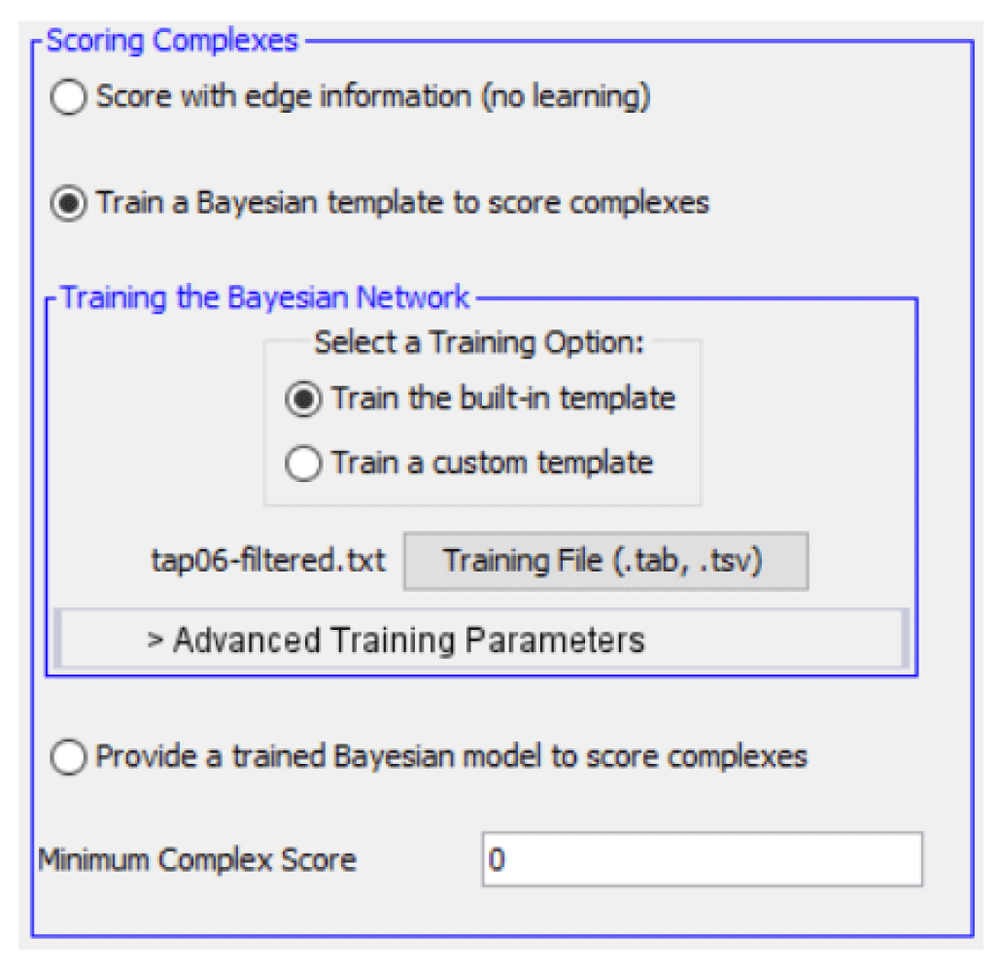
SCODE offers two scoring options to the user. The first does not perform learning, while the second option does (by training a Bayesian model).
The first scoring option performs a simple calculation based on the mean weight among edges in the proposed protein cluster. It does not require any information regarding known complexes and does not perform learning. This option may be used as a reference for comparing the complexes produced by the second scoring method.
The second scoring option, relying on supervised learning, calculates scores using a likelihood equation that is generated based on a trained Bayesian network. It reflects the conditional probability distribution that each feature is demonstrated in a complex versus the distribution for a non-complex. The sample Bayesian network from Figure 2 would use the following calculation to determine the likelihood that a candidate protein cluster, x, represents a true complex, given the prior probability that x is a complex (P(x1)) or a non-complex (P(x0)):
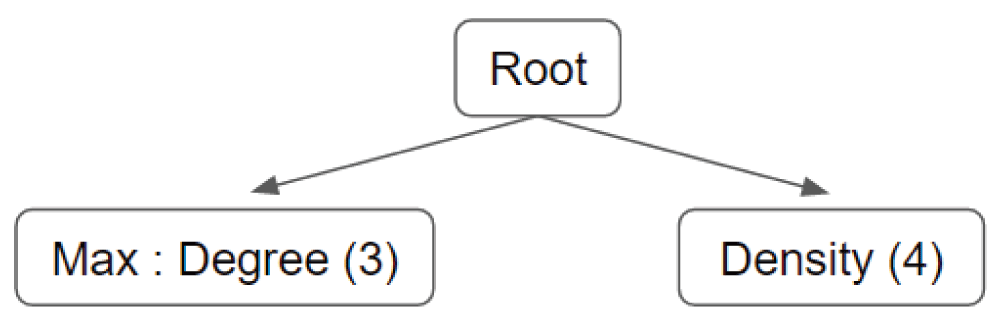
The root indicates whether the candidate cluster is a complex or not.
Here P(f1|x1) denotes the conditional probability that feature f1 is observed given that x is a complex (x1). P(f1|x0) indicates the conditional probability that feature f1 is observed given that x is not a complex (x0). This equation may be extended to any naive Bayesian network, with f1…fn representing the features of the model:
Bayesian networks (BNs) are directed acyclic graphs (DAGs) that describe the conditional dependency relationships among a set of nodes representing random variables. In SCODE, Bayesian networks are used to describe the joint probability distribution of the features describing complexes, as well as the joint probability distribution of the features describing non-complexes in the PPI graph. After training the networks to calculate each of the conditional probabilities in Equation 1, these networks are used to score complexes that are discovered during the search phase.
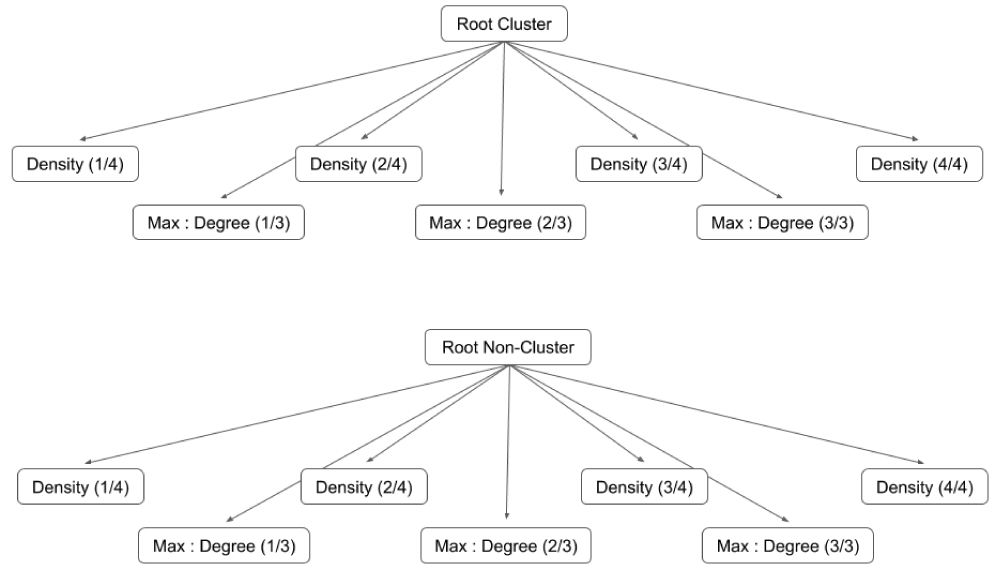
A feature describes a particular property of a candidate complex, such as size or density. Each node in the trained Bayesian model represents a discrete value, or subset of values, of a feature. For example, a binary feature is represented in the trained model using two nodes, with one node for each of its two possible values. A feature with continuous values is discretized by subdividing its range into a set of equally-sized bins. This representation of the Bayesian model allows the conditional probability of each discrete value (or range of values) of a feature to be stored in the Cytoscape network as an edge table attribute. However, as Figure 3 demonstrates, this structure is incredibly verbose and requires multiple nodes to separate the bins for a single feature. We instead provide a simpler way for users to design a Bayesian template (an example is shown in Figure 2).
Users may define custom Bayesian templates using Cytoscape’s native graph-building or importing tools. These templates must use a specialized syntax for naming nodes, wherein each node other than the root (labeled ‘Root’) specifies a feature. The syntax for a feature is as follows:
"Statistic" is an optional prefix that indicates an operation to perform on the values of a feature (such as taking the max, mean, or median). A statistic must be applied to a feature that produces a list of values rather than a singular value. Applying the statistic results in a single floating-point value, which may then be assigned to the appropriate bin. The available statistics include: Mean, Median, Count, and ordinals such as 1st, 2nd, 3rd, etc.
For example, Figure 2 illustrates a simple Bayesian network with two features that are conditionally independent given the root node. The feature labeled "density" may have continuous values in the range (0, 1], divided equally among 4 bins. In the second feature, "degree" represents the number of neighbors of each node in a candidate cluster. Applying the "max" statistic produces the highest degree among all the nodes in a cluster. The range of this feature is (0, n], where n is the highest degree of any node in the positive training data.
Once its features are defined, the template is trained using a set of user-provided positive training complexes and a set of randomly-generated negative training clusters. First, two nearly-identical representations of the Bayesian template are created, with the root node of the first labeled ‘Root Cluster’ and the root node of the second labeled ‘Root Non-Cluster’. A separate node is then created for each bin of each feature, resulting two networks whose respective sizes are, identically, the sum of the number of bins for each feature plus the root node. Figure 3 illustrates the two networks produced after training the template in Figure 2.
The positive Bayesian network (‘Root Cluster’) is trained using the positive complexes provided by the user, and the negative Bayesian Network (‘Root Non-Cluster’) is trained using the randomly-generated negative protein clusters. In both training procedures, the program records the frequency at which the values representing each node in the Bayesian network are encountered among the training clusters; this translates to the frequency for each feature bin.
By the end of training, the node frequencies are used to encode the conditional probabilities, P(fb|RootCluster) and P(fb|RootNon − Cluster) for bin b of feature f, as follows:
We may then use Equation 1 to produce a log-likelihood score for a candidate cluster based on the conditional probability of each feature bin in both the positive and negative Bayesian networks. Conditional probabilities on the root node are maintained as properties of the edges in the returned Cytoscape network, which represents both halves of the trained Bayesian model; in doing so, trained models may be saved and recycled for later use in Cytoscape session files.
The search phase returns a set of predicted complexes and their associated likelihood scores, which may be visualized and analyzed independently using Cytoscape tools. SCODE also provides a tool for evaluating discovered complexes against a user-provided set of known complexes in the network, allowing the program to quantify its overall performance.
To perform the evaluation, the user must supply a file containing a list of complexes known to exist in the PPI graph. These complexes may include the positive examples used to train the Bayesian network, but they will be filtered out when calculating the evaluation score.
The evaluation provides two metrics: recall and precision. Recall measures the ability of the program to discover complexes from the known set, while precision measures the accuracy of the discovered complexes.
When comparing a predicted complex against a known complex,
• Let A be the number of proteins only in the predicted complex
• Let B be the number of proteins only in the known complex
• Let C be the number of proteins in both the predicted and the known complex
A predicted complex is said to have identified a known complex3 if
Here p is a hyperparameter specified by the user. Recall and precision are calculated as follows:
SCODE is organized among three packages. The statistic package contains implementations of the abstract class ’Statistic’, which specifies methods for operating on a list of feature values and calculating the overall range of those values for binning. A second package, feature, contains implementations of the abstract FeatureSet class. FeatureSet specifies methods for getting feature values, applying statistics to them, and returning the bins for a complex. The list of statistics that is applied to a feature is stored as a protected member of the FeatureSet class.
The third package contains the remaining code for gathering input and executing the search, scoring, and training tasks. The entry point to SearchTask and TrainingTask creation is in SupervisedComplexTaskFactory. TrainingTask initiates the process of loading the appropriate template network or trained model network, which is then used to create the internal representation of the Bayesian network in the SupervisedModel class. SupervisedModel represents both the positive and negative Bayesian graphs via the Graph class, which also specifies methods for training the graph and scoring candidate complexes during search.
From the initial searchTask, an IsaSearch object is created to divide the starting nodes among separate threads and to begin searching on each one. The core search algorithm is located in the SeedSearch class, where candidate complexes are expanded and scored. Complexes are represented internally as Cluster objects and returned as CySubNetworks once the search has been completed.
Taking advantage of its new modular architecture, SCODE requires Cytoscape version 3.2 or above to operate. Upon launch, SCODE accepts user input and displays a summary of the results from the SCODE tab in the Control Panel. Mirroring its pipeline, SCODE’s interface is divided into subsections for each of the search, scoring, training, and evaluation tasks.
The user may adjust the following input parameters for the search stage:
• Variation of Simulated Annealing : The program offers three variants of the Iterative Simulated Annealing algorithm: ISA, Sorted-Neighbor ISA, and Greedy ISA.
ISA : Fastest performance but tends to produce low-scoring complexes.
Greedy ISA : Slower than ISA, but tends to produce more, larger, higher-scoring candidate complexes.
Sorted-Neighbor ISA : Slower than ISA and sometimes slower than Greedy ISA, depending on the density of the PPI graph. Tends to produce higher scoring complexes.
• Search Limit : Specifies the maximum number of iterations of the search.
• Initial Temperature : Sets the starting temperature of the search. Higher temperature increases the likelihood that a protein will be added to the complex.
• Temperature Scaling Factor : The rate at which the temperature decreases over time.
• Overlap Limit : The maximum proportion of nodes that two distinct complexes may share.
• Use Seeds From File : Optional; the user may select an external file containing a list of seed nodes from which to begin the search.
• Number of Random Seeds : Allows the program to randomly select the specified number of seed nodes.
• Number of Results to Display : The number specified here will be the number of results visible at the end of the search.
The scoring section accepts the following parameters:
• Minimum Complex Score : When the results are shown to the user, only candidate complexes with scores equal to or greater than this input will be displayed.
The training section accepts the following parameters:
• Cluster Probability Prior : The prior probability, P(x1), that a group of proteins forms a complex.
• Generate Negative Examples : Specifies the number of negative training examples that the program will randomly generate.
• Ignore Missing Nodes : During training, if one of the proteins in a positive training example cannot be found in the PPI network, selecting this option will allow the program to disregard the training example.
The training section takes additional input parameters relating to the Bayesian network. The first set of parameters specifies the Bayesian network itself. The user may either use a default model provided by the application, or a custom Bayesian network that has been constructed using Cytoscape. The custom network may be either trained or untrained; in the latter case, the user must provide an input file containing a set of known complexes that will be used to train the model. Trained models can be saved and loaded from Cytoscape session files, which store networks for later use.
SCODE includes a number of predefined features for building Bayesian templates, enumerated below. The features of the built-in Bayesian template are provided in Table 2, but are not exhaustive. In the following descriptions, G indicates a PPI graph with edges, E, and nodes, N:
• Complex Size : Takes the length of a list of complexes of size |N|.
• Clustering Coefficient : How many triangles contain the node n as a vertex. Calculated using the equation CCn = 2ek/(kn * (kn − 1)), where kn is the number of neighbors of n and ek is the number of edges between those neighbors.
• Degree : For a node in N, the number of edges connected to that node.
• Degree Correlation : For a node in N, the average number of neighbors among its adjacent nodes.
• Density : The ratio of the number of observed edges to the number of possible edges in G: D = |E|/|Ep|.
• Density at Cutoff : The same as above, once edges with a weight below the cutoff are removed from the set of observed edges.
• Edge Weight : A measure of the strength of the interaction between a pair of nodes. Must be provided as an edge table column in the PPI network.
• Edge Table Column : Produces a vector of values from a Cytoscape graph table’s column for each edge in the graph.
• Edge Table Correlation : Produces a set of vectors from a set of columns in a Cytoscape table and returns a list containing the correlation coefficients for each pair of vectors.
• Node Table Column : The same as the edge table feature, but applied to each node.
• Node Table Correlation : The same as the edge table correlation feature, but applied to nodes.
• Singular Values : The singular values for each graph correspond to the graph’s shape. For instance, A linear graph’s values will consistently differ from those of a clique.
• Topological Coefficient : For a node n, the topological coefficient measures the connectivity of n’s neighbors, where f (a, b) is the number of neighbors in common between neighbors a and b, while kn is the number of neighbors: TCn = avg(f (a, b))/kn
We demonstrate the app using training/evaluation datasets constructed from two sources: the CYC2008 catalogue of manually curated protein complexes4, and the TAP06 dataset of complexes screened using affinity purification and mass spectrometry5, which were each filtered to a random sampling of 50 complexes with 4–6 nodes. Both files are supplied in a tab-separated format.
The graph on which search is performed features 396 proteins (those featured among the CYC2008 and TAP06 complexes) and 5141 edges. The set of interactions in this graph is generated from the STRING database of known and predicted protein-protein interactions6. The graph is also in tab-separated format and must be loaded into Cytoscape as a network; the network may then be saved in a session file (.cys) for later use. All of the data used in this demonstration can be found online and is provided below under ‘Data Availability’.
At the end of the search, discovered complexes will be returned as Cytoscape networks under the ‘Network’ tab in the Control Panel.
For this demonstration, we employ the parameters shown in Figure 4. The number of random seeds matches the number of nodes in the graph. This number, to some degree, dictates the likelihood of identifying complexes of good quality. With a low starting seed count in a large PPI graph, the probability of beginning the search from a protein that is a member of a "true" complex is low. For the purposes of our evaluation, we provide the ideal conditions for the search to return true complexes. This includes using Greedy ISA so that all nodes neighboring a complex are scored before selecting one for expansion.
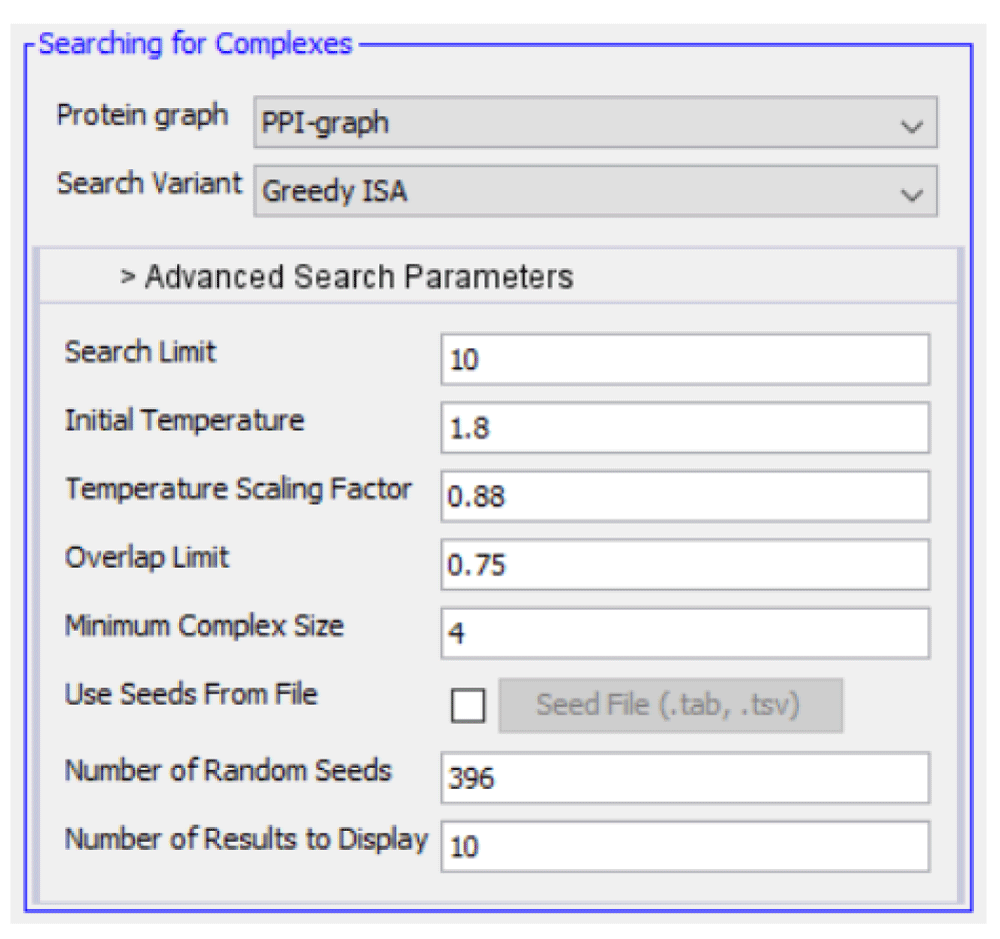
We choose to employ the built-in Bayesian template for training, with features shown in Table 1. Since the PPI graph is under 500 nodes, we use a prior probability of 0.5 for clusters, and generate 50 negative examples. Two rounds of training and search are performed, the first using the abridged set of CYC2008 complexes for training and the TAP06 complexes for evaluation, and the second using the reverse (with all other parameters held constant). The minimum complex score is set to 0.
Once the search is complete, the top M results (specified under the search parameters) are displayed under the ‘Network’ tab in the Cytoscape Control Panel. All protein clusters produced by the search, including but not limited to those displayed, are considered during evaluation. The Evaluation section appears directly below the Scoring section after ‘Analyze Network’ is clicked (Figure 6).
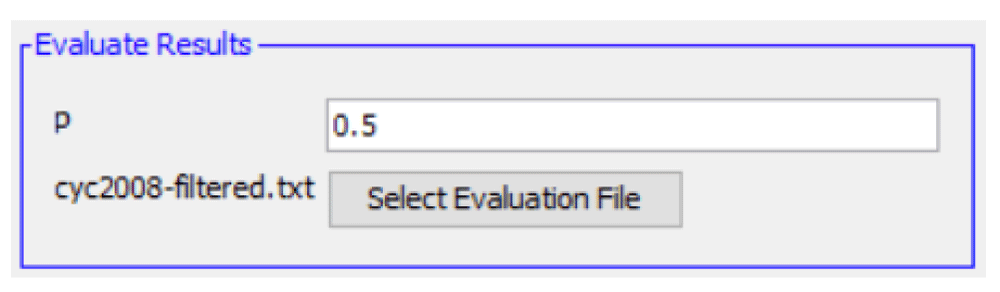
We supply p=0.5 for Equation 3, such that all predicted complexes with a majority of proteins from a known testing complex are said to have "recovered" that complex. After clicking "Evaluate Results", the evaluation scores for recall and precision appear in a dialog (Figure 7).

Evaluation scores using CYC2008 to train and TAP06 to test (a) or TAP06 to train and CYC2008 to test (b).
SCODE expands the detection of protein complexes in weighted PPI networks by applying a supervised learning algorithm with a set of known training complexes. Users may discover topologically non-traditional protein complexes by leveraging more information about the features of its PPI graph. The Bayesian network encodes the desired characteristics of complexes beyond density and is used to score the likelihood that a candidate discovered during a search of the PPI network represents a complex. Each version of the ISA search heuristic discovers complexes of varying quality and size, in accordance with the degree to which the algorithm exhausts the search space.
F1000Research: Dataset 1. Demo Using CYC2008 and TAP06 Complexes, 10.5256/f1000research.9184.d1288788
1. Software available from:
2. Latest source code: https://github.com/DataFusion4NetBio/Paper16-SCODE
3. Archived source code as at time of publication: http://dx.doi.org/10.5281/zenodo.571639
4. Software license: MIT License (https://opensource.org/licenses/MIT)
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)