Keywords
human endocrine pancreas derivation, Induced pluripotent stem cells, CRISPR-Cas9, maturity-onset diabetes of the young, Wolfram syndrome,
human endocrine pancreas derivation, Induced pluripotent stem cells, CRISPR-Cas9, maturity-onset diabetes of the young, Wolfram syndrome,
Type 2 diabetes (T2D) is a global health burden. Given that more than 415 million individuals are currently affected and that the incidence is predicted to rise faster than the adult population growth rate1, it could be argued that our current preventative and therapeutic strategies against this disorder are inadequate.
Understanding T2D pathophysiology is inherently difficult because of its complex aetiology; an individual’s disease risk is based on a combination of genetic, epigenetic, environmental, and lifestyle risk factors2,3. However, the last decade or so has seen a transformation in our understanding of the genetic basis of this disease; through large-scale international collaborations and DNA samples from hundreds of thousands of individuals, common and rare variant association studies have identified more than 100 genomic loci influencing T2D susceptibility4,5. Also, for T2D, and unlike many other complex genetic disorders, we have a good handle on the tissue driving pathogenesis; despite perturbations to both insulin secretion and sensitivity, multiple studies place pancreatic islet dysfunction at centre stage in terms of disease susceptibility and progression6–8.
Despite this wealth of information, our ability to go from genetic signal to mechanism (and even therapeutic target) has progressed at a pace far slower than that of the initial discoveries of these disease susceptibility loci. Why?
Multiple factors underlie the difficulties in biological interpretation of genome-wide association study data. Firstly, we need to know which transcript(s) are driving the phenotypic signal. This has formed a huge stumbling block for researchers as (i) extensive regions of linkage-disequilibrium mean that most associated loci harbour many genes and transcripts, (ii) many signals lie within poorly annotated, non-coding regions of the genome (although efforts to map the ‘islet regulome’ are beginning to bear fruit9–12), and (iii) the modest effect sizes of disease-associated variants make functional interrogation of risk versus non-risk alleles problematic (odds ratios are usually between 1.1 and 1.44,5).
Secondly, far and away one of the biggest challenges has been the lack of appropriate human islet cell models for study. Until very recently, this was limited to animal models and rodent insulinoma cell lines, which present numerous challenges; there are multiple instances in which human diabetic phenotypes are not recapitulated in the analogous murine model of gene haploinsufficiency13–24, and differences in islet architecture, ion channel composition, nutrient sensitivity, and other physiological parameters25–31 limit the functional inferences that can be made from rodent-derived data. Human islet isolation programmes and the subsequent availability of this tissue for research purposes have gone some way to alleviate this bottleneck, as has the recent generation of human beta-cell lines from pancreas explants32,33, although these latter cells are only just beginning to be characterised34.
Thirdly, despite increasing access to human islets and cell lines, many technical constraints remain: (i) human islets are heterogeneous in terms of donor genotype and function/viability after surgical extraction, (ii) the restriction of islet isolation programmes to adult donors limits study to mature cells, (iii) human beta-cell lines represent only a single islet cell type, and (iv) low recombination rates and an inability to expand single clones make genomic manipulation via site-specific nucleases challenging.
This article will focus on one of the most exciting emerging fields in diabetes research at present: human endocrine pancreas derivation in a dish. The utilisation of state-of-the-art in vitro differentiation techniques to turn human pluripotent stem cells into those of the islet lineage35–41 allows researchers to sequentially generate definitive endoderm cells (expressing SOX17 and FOXA2) through to pancreatic progenitors (PDX1- and NKX6.1-positive), all the way to cells expressing insulin, glucagon, and islet transcription factors regulating mature cell function (MAFA).
This model system has broad application in many areas of islet biology and diabetes research. Firstly, it can be used as a platform for drug discovery efforts aimed at increasing functional beta-cell mass, and importantly, one which is without many of the ethical, legal, and practical considerations surrounding the routine use of human tissue (both foetal and adult). Induced pluripotent stem cells (iPSCs) specifically bypass the need for embryonic tissue as they can be generated by reprogramming any somatic cell42,43. Secondly, the ability to further mature these cells in vivo, and to phenotypically correct diabetes in immunocompromised mice38–40,44–49, also shows the translational potential of such cells, with analogous clinical trials beginning to take place in humans50. Both of these areas have been reviewed extensively elsewhere51–56. Instead, the rest of this article will focus on the potential of stem cell-derived islet-lineage cells in disease modelling, in particular how they can be manipulated with genome editing tools such as CRISPR-Cas957,58, so as to accurately recapitulate the genomic, developmental, and mature cell perturbations underlying T2D pathogenesis59 (Figure 1).
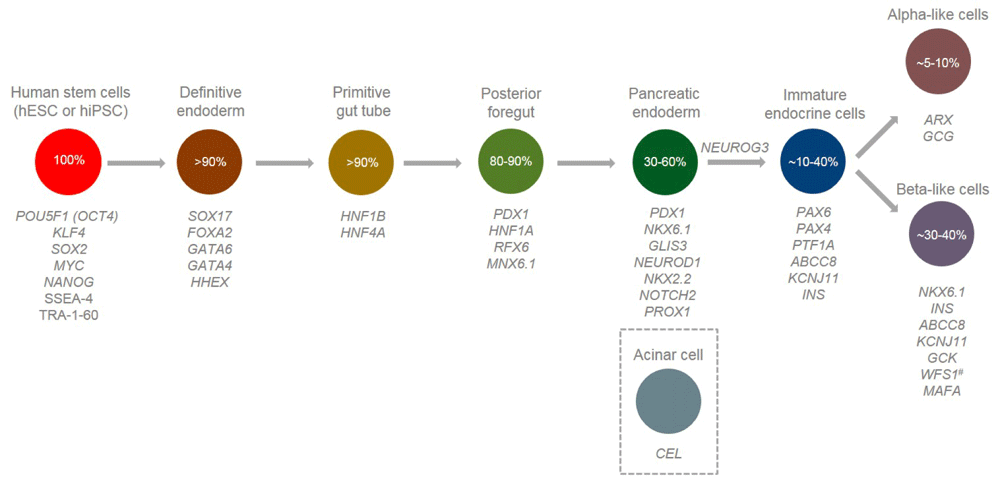
Circles represent discrete developmental stages, with derivation efficiency estimates also shown59. Genes discussed in this article are listed according to the developmental stage at which they are first expressed and any subsequent stages where they perform important biological functions or are crucial for cell identity (#except WFS1 which is expressed at all stages; however, the diabetes observed in patients with Wolfram syndrome is believed to result from selective beta-cell loss via apoptosis76). CEL is expressed in acinar cells, which differentiate from multipotent pancreatic progenitor cells and subsequently exocrine progenitor cells (not depicted in the figure). hESC, human embryonic stem cell; hiPSC, human induced pluripotent stem cell.
Recent methodological advances in endocrine pancreas differentiation have promoted formation of mono-hormonal cells with function similar to (but not quite yet the same as) that of human islets38–41. However, variation in line-to-line differentiation efficiencies60–62 coupled with an inability to make fully mature cells63 has so far limited disease modelling to monogenic diabetes caused by highly penetrant, large-effect mutations.
One of the first proof-of-principle studies64 generated iPSC lines from individuals with maturity-onset diabetes of the young (MODY) by using a polycistronic lentiviral vector overexpressing the so-called ‘Yamanaka factors’ (POU5F1 [OCT4], KLF4, SOX2, and MYC), these needed for somatic cell reprogramming to pluripotency42. This included lines from patients with mutations in endocrine pancreas developmental transcription factors (HNF1B, HNF4A, and HNF1A), as well as those with perturbed enzymes governing glucose-stimulated insulin secretion (GSIS) in mature cells (GCK), and even exocrine pancreas function (CEL). Regardless of mutated gene, all lines were shown to fulfil basic iPSC quality control: expression of pluripotency genes via fluorescence-activated cell sorting (OCT4, SOX2, NANOG, SSEA-4, and TRA-1-60), spontaneous teratoma formation upon transplant into immunocompromised mice (cells capable of generating all three germ layers), and a diploid ‘stable’ karyotype64. Another study aimed at generating iPSCs from patients with HNF1A-MODY65 again produced cells passing basic pluripotency QC, and which were able to differentiate from embryoid bodies, into those expressing insulin and glucagon. Of note here is that these hormones were not present at levels comparable to those seen in other studies39,40, perhaps reflecting the quite different in vitro differentiation strategies employed. Likewise, the inability of these cells to form teratomas spontaneously in vivo suggests that reprogramming to full pluripotency may not have been achieved.
Other diabetes iPSC models have focussed on characterising cellular dysfunction apparent within mature islets, making endocrine pancreas differentiation essential for phenotyping patient-derived cells. Individuals with heterozygous GCK mutations have a mild phenotype whereby fasting plasma glucose levels are marginally elevated (6 to 8 mmol/L) because of a higher threshold for GSIS, which is governed by altered beta-cell glucose uptake and glycolytic flux66,67. Directed differentiation of iPSCs from patients with GCK-MODY down the islet lineage occurred with an efficiency comparable to that of control cells, with the only observable defects mirroring patient phenotype (elevated GSIS set-point), thus validating this as a physiologically representative model for studying monogenic GCK mutations68.
iPSC models have also been generated for syndromic diabetes disorders, such as Wolfram syndrome. This disorder is caused by mutations in WFS169, with patients suffering from multi-organ dysfunction, including diabetes, optic atrophy, and neurodevelopmental defects70. Such a broad phenotype reflects the multi-tissue expression of WFS1, with the encoded Wolframin protein performing vital roles in endoplasmic reticulum (ER) Ca2+ homeostasis71–73 as well as alleviating ER stress in cells with high translational load74, such as those with secretory function75. This is thought to explain the childhood-onset diabetes in these individuals, with post-mortem study of Wolfram syndrome pancreases suggesting selective beta-cell loss via apoptosis76. Directed differentiation of iPSCs from patients with Wolfram syndrome down the islet lineage showed that these cells had elevated levels of chemically induced ER stress, which resulted in translational stasis and decreased insulin processing and content. Likewise, in vivo maturation of patient cells showed that grafts declined in function much more rapidly than control cells, perhaps reflecting enhanced apoptosis77.
Importantly, the cellular dysfunction observed in both diabetes iPSC-derived models was corrected via genetic (zinc finger nuclease68) or chemical (4-phenylbutyric acid77) means. This phenotypic correction is fundamental in assigning causality to the studied mutation of interest, particularly as large-scale sequencing studies are continuing to identify previously reported ‘disease-causing’ mutations in unaffected individuals within the general population, leading to continued revision and reduction of penetrance estimates78. Likewise, comparing patient lines to isogenic controls removes any differentiation efficiency or phenotypic effects driven by factors extrinsic to the particular mutation of interest, including reprogramming efficiency and epigenetic or sequence variation (or both) in the donor genome60–62.
A methodological advance which has revolutionised the ease at which we can generate isogenic control lines is the expansion of site-directed nuclease, so-called ‘genome editing’ technologies, from zinc finger nucleases79,80 to TALENs (transcription activator-like effector nucleases)81–85 and more recently to CRISPR-Cas957,58,86–100. The most popular of these editing methods, CRISPR-Cas9, exploits a bacterial innate immune system response to pathogens, whereby the Cas9 endonuclease is targeted to invading phage DNA by a sequence-specific guide RNA molecule101. In recent years, manipulation of this system so that it can target eukaryotic (specifically mammalian) genomes has allowed its full translational potential to be realised57,58,102. The ability to target more or less any sequence in the human genome for gene knockout via non-homologous end-joining103, nucleotide-level manipulation via homology-directed repair104,105, or larger recombination events to generate reporter lines90,106 or even bring into close proximity mediators of gene expression (such as activators or repressors tethered to modified Cas9 protein)86–88,92,93,107 means that every type of genetic perturbation is theoretically possible. Use of this technology has also extended into simultaneous targeting of multiple genes57,87,88,98–100 as well as inducible89 and epigenome-modifying91,108 systems.
Accordingly, CRISPR-Cas9 and other site-specific nucleases are a very attractive tool for the generation or correction (or both) of diabetes-relevant mutations in human stem cell-derived models, stem cells being particularly amenable to this technology because of their clonal nature and highly recombinogenic genome. Both gene knockout via Cas9-induced indels109 and doxycycline-inducible gain-of-function transgenes (targeted to the AAVS1 safe harbour locus using TALENs110) have been used to definitively establish the role of NEUROG3 in human pancreas development. Whilst Neurog3 is essential for murine pancreas development and derivation of all islet cell types111–113, individuals with homozygous NEUROG3 mutations retain some islet function114–116. Complete gene knockout showed that NEUROG3−/− cells could not mature past pancreatic progenitors into endocrine pancreas; however, with graded perturbation to gene dosage via small hairpin RNA (shRNA), as little as 10% residual NEUROG3 activity still led to some islet hormone-positive cells109. These data are directionally consistent with analogous experiments whereby inducible NEUROG3 overexpression in human embryonic stem cell (hESC)-derived pancreatic progenitors leads to increased numbers of endocrine pancreas-like cells expressing INS, NKX2.2, NEUROD1, and other relevant islet transcription factors110. Drastically reduced NEUROG3 levels are therefore sufficient for the development of human islets, an effect not recapitulated in mice.
Although many reports have begun to emerge of mutation introduction or correction via homology-directed repair in both control and patient-derived cell lines, these remain as yet unpublished, perhaps reflecting the low efficiency of this technique and repeated cleavage of repaired sites117, alongside the additional scrutiny of these experimental techniques in terms of off-target effects118,119.
Patient-derived iPSCs facilitate study of the precise mutational mechanisms underlying an individual’s diabetes risk and progression; however, their use so far has been limited to monogenic disease. Although we may not yet have phenotypic resolution to assay dysfunction underlying more complex disease, the ability to generate cellular models of islet development opens up a whole new avenue of investigation for T2D pathogenesis59.
We know from studying monogenic diabetes and pancreatic agenesis that there is substantial overlap between the genes causing these phenotypically severe Mendelian disorders and those harbouring more common and incompletely penetrant variants predisposing to T2D risk4,120. It follows that within these cellular pathways, the extent of perturbation dictates when diabetes presents: either in utero/early life if severe or much later as T2D if more subtle.
At the extreme end of this scale is pancreas hypoplasia or even lack of a pancreas completely (agenesis). Haploinsufficiency for GATA6 is the most common cause of pancreatic agenesis in humans14. Individuals with this haploinsufficiency may also experience cardiac or gastrointestinal abnormalities, reflecting the role of GATA6 in organogenesis for multiple tissues. As phenotypic presentation of GATA6 mutation carriers varies (some individuals experience dysfunction in only a subset of these tissues), a potential redundant role for the related transcription factor GATA4 has been proposed in humans. This hypothesis is well established in mouse development121,122 but continues to be the subject of debate in humans, despite the identification of individuals with neonatal diabetes (one with pancreatic agenesis) resulting from heterozygous GATA4 mutations20.
Biallelic inactivation of RFX6, a key transcription factor in gut- and pancreatic-endoderm specification, causes both neonatal123–125 and childhood-onset diabetes126, with phenotype severity correlating with loss of RFX6 gene dosage, and subsequently islet cell development/hypoplasia126. An elegant CRISPR-Cas9 hESC knockout study showed that loss of RFX6 alters or delays pancreatic progenitor formation through perturbed PDX1 induction110, thus implicating RFX6 in the regulation of both foetal and adult islet cell function (in which it helps maintain mature beta-cell identity127,128). Heterozygous mutations in HNF1B129, a gene switched on within cells in the primitive gut tube where it is responsible for regional gut specification and branching morphogenesis as well as later cell fate decisions in multipotent pancreatic progenitors130, cause MODY131,132, pancreatic hypoplasia/agenesis133–136, and renal abnormalities137–139.
GATA6 and HNF1B map to genomic loci implicated in later-onset diabetes4,120; therefore, understanding their role in foetal and adult human islets is crucial for investigating T2D pathogenesis. Because mice haploinsufficient for Gata6, Gata4, and Hnf1b do not have diabetes15–17 and with homozygous knockouts causing embryonic lethality140–142, dual developmental and adult characterisation would not be possible without human cell models representative of both time points.
The severity of a diabetes phenotype may be influenced, in part, by the temporal expression pattern of a mutated gene. For example, one of the downstream targets of HNF1B is GLIS3, a zinc finger transcription factor involved in regulating the transient spike in NEUROG3 expression important for endocrine fate commitment130. Although GLIS3 mutations have been shown to cause neonatal diabetes and T2D in humans, these same individuals do not experience pancreatic agenesis143, and this fits with the later expression of GLIS3 (versus HNF1B) in the foetal pancreas. This suggests that these individuals are able to make some endocrine pancreas tissue and that disease pathology results from insufficient insulin secretion from a reduced functional beta-cell mass potentially both in utero and in adult life. Analogous observations have been made for individuals with mutations in the foetal pancreatic transcription factors PAX6144, NEUROD1145, NKX2.2146, and MNX1146,147.
In a similar vein, heterozygous mutations in other genes important for islet progenitor function can cause the milder phenotype of MODY; this is characterised by onset of non-insulin-dependent diabetes before 25 years of age148. Mutations in HNF family members HNF4A and HNF1A are the most common cause of MODY in Europeans149–153, and these genes also map to genomic regions associated with T2D risk4,120. Whilst both disorders could result from defective insulin secretion from mature islets (the two transcription factors regulate genes governing GSIS24), they also perform distinctive roles in the foetal pancreas, as dictated by discrete spatiotemporal expression patterns for each of the multiple HNF4A and HNF1A transcript isoforms154–156. Studying both HNFs in foetal versus adult tissue has also shown big differences in post-translational regulation; in adult islets these two HNF transcription factors regulate expression of each other and themselves157 whereas only HNF4A mutations have been shown to cause the more severe phenotype of neonatal diabetes, suggesting that this gene has a more dominant role in foetal pancreas development155. The association of HNF4A variants with macrosomia and hypoglycaemia in neonates158 also suggests that perturbations to this gene transiently increase foetal insulin secretion, a phenomenon not observable if studying (i) adult islets alone (as HNF4A mutations cause the opposite phenotype of beta-cell dysfunction and hyperglycaemia153) or (ii) rodent pancreas (Hnf4a+/− and Hnf1a+/− mice are phenotypically normal13,18,24). Accordingly, understanding the temporal relationship between HNF4A gene dosage and insulin secretion is fundamental to managing pregnancy as well as neonatal and young-onset diabetes and T2D.
Irrespective of a previous implication in Mendelian diabetes, knowing the developmental expression pattern of genes mapping to T2D-associated regions of the genome can also help refine likely effector transcripts at these loci, particularly considering the well-established role of islet dysfunction in the progression of this disease6–8. HHEX, NOTCH2, and PROX1 map to T2D loci containing multiple putative effector transcripts and potentially causal variants4,120. Although none of these genes harbour mutations implicated in monogenic diabetes, strong candidacy for their role as effector transcript comes from their importance in endocrine pancreas development: HHEX regulates ventral pancreas organogenesis159, NOTCH2 is involved in fate decisions of pancreatic progenitors160, and PROX1 marks pancreatic progenitor cells in the endoderm (later becoming specific to NEUROG3-positive cells)161. Thus, using human models of endocrine pancreas differentiation to understand how subtle perturbations to these genes during development may impact upon risk of diabetes in later life is fundamental to the functional characterisation, and consequent assignment of variant/transcript causality, at these T2D-associated genomic loci59.
This same principle can be applied to disentangling disease-associated genetic perturbations mapping to non-coding regions of the genome. As many islet enhancers are tissue-specific162, and with studies in stem cell-derived endocrine pancreas-lineage cells also showing these and other regulatory marks to be developmental stage specific too163, it follows that characterisation of non-coding regions harbouring disease-associated genetic variations is possible only if developmental pancreas cell models are employed. A good example of this approach comes from a recent study of multiple consanguineous families with recessive pancreatic agenesis of unknown aetiology164. All affected individuals were absent of coding mutations in previously established pancreatic agenesis genes (GATA614, PTF1A165, and PDX1166,167) and accordingly were subjected to whole genome sequencing. Homozygosity mapping showed that no biallelic coding changes co-segregated with disease. Extended analysis into non-coding regions of the genome showed that multiple affected individuals harboured biallelic mutations in a 400-base pair sequence about 25 kB downstream of PTF1A, a transcription factor mediating early pancreas specification from the foregut168. ChIP-seq in hESC-derived pancreatic progenitors showed that this region overlapped binding sites for the foetal pancreas transcription factors FOXA2 and PDX1 as well as an H3K4me1 active enhancer site. Enhancer activity was shown to be tissue- and developmental stage-specific and was abolished upon introduction of the agenesis mutations164. As PTF1A maps to a locus associated with T2D4,120, it follows that similar developmental enhancers may also be important in adult-onset disease.
Although as diabetes researchers we can put a large emphasis on understanding insulin secretory defects, aberrant glycaemia can also result from dysfunction in other islet cell types.
Because differentiated stem cells make cells positive for all islet hormones39,40, one can use the same systems to study aberrant glycaemia resulting from perturbations in non-beta cell types. Diffuse congenital hyperinsulinism in infancy (CHI) is characterised by insulin over-secretion despite hypoglycaemia169. Mutations in the ATP-sensitive islet potassium channel subunit genes ABCC8 and KCNJ11 are the most common cause of CHI; the unregulated closure of this channel is thought to result in sustained insulin release170,171. However, study of pancreas tissue from 10 individuals with KCNJ11-mediated CHI showed that functional beta-cell mass was maintained as constant since, despite increased proliferation, a concomitant elevation in cell type-specific apoptosis was also observed172. Intriguingly, and consistent with the disorganised islet architecture observed in Kcnj11 knockout mice21, the human CHI islets had downregulated PAX4 and ARX levels (the latter transcription factor specific to alpha cells173) as well as elevated NKX2.2 expression (particularly in delta cells, 10% of which also demonstrated nucleomegaly)172,174,175. Consistent with the use of somatostatin analogues in the treatment of some CHI cases169, these data suggest that alteration of multiple endocrine pancreas cell lineages (not just beta cells) is driving phenotype172. Despite disorganised islets, Kcnj11 and Abcc8 knockout mice do not exactly recapitulate the phenotype of human CHI21,22, making the further investigation of this disorder in stem cell-derived endocrine pancreas models attractive.
This review highlights the need for human, physiologically relevant cell models which accurately recapitulate both foetal and adult islet function for interrogation of diabetes pathogenesis. Although a lot of our knowledge regarding pancreas development has come from studying the mouse, there are many cases in which murine models fall phenotypically short and so translating genetic signals into disease mechanisms is limited. The huge advances that have been made in differentiating human stem cells (both embryonic and induced pluripotent) into all cell types of the developing endocrine pancreas have transformed how we are able to characterise disease-causing and -associated genetic perturbations. However, although we are now able to make endocrine pancreas-like cells with some islet function, it is important to temper expectations and remember that we are still some way from making the perfect beta cell. Although the most recent studies from leading labs report glucose-responsive insulin secretion and Ca2+ channel activity39,40, this function does not fully recapitulate that of human islets. Accordingly, we as a field must make an effort to standardise phenotyping assays and subject them to the same scrutiny as that used to interrogate primary tissue. Efforts to deposit functional176 and omics-level59 data for both primary tissue and stem cell-derived endocrine pancreas-like cells are helping researchers generating their own pancreas-in-a-dish to compare, contrast, and truly evaluate their model systems. Once this methodological standardisation is achieved, we can collectively increase the complexity of our routine phenotyping of parameters such as hormone secretion and ion currents and move towards physiologically relevant doses of mixed nutrient stimuli, amongst other assays.
Regardless of these current functional bottlenecks, coupling stem cell-derived endocrine pancreas-like cells with the excitement of genome editing technologies places diabetes researchers in an extremely powerful position of novel biology discovery and genetic signal validation. Armed with these new experimental tools, one can start probing more complex forms of the disease such as T2D59 and, with a pluripotent cell type, model the complex multi-organ dysfunction occurring in cells derived from the same patient. The dream of a true ‘personalised medicine’ approach to diabetes is in our midst.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)