Keywords
T-cell, T-cell signalling, mTOR
A recap of “T-cell activation” and the checkpoint inhibitor breakthrough
Checkpoint inhibitors represent a breakthrough treatment class for patients with cancer1–3 and derive from investing in basic science and fundamental questions of T-cell signaling. What are the roots of this revolution and how does it relate to signaling inside T cells? On conceptual grounds, Bretscher and Cohn hypothesized that T-cell activation should require two signals4. Myriad studies established the principle that T cells would be activated through an antigen receptor and that this T-cell antigen receptor (TCR) was stimulated by encounter with a cell whose surface bears a specialized protein encoded in the major histocompatibility complex (MHC) combined with a suitable peptide5,6. TCR interactions with an appropriate MHC-peptide complex, however, could yield a state of unresponsiveness when an arcane chemical fixative was used to analyze antigen presentation7. These findings implied that TCR stimulation was not enough; a co-stimulatory molecule was required, consistent with the model by Bretscher and Cohn. A race to elucidate the mechanism culminated in identification of the CD28 co-stimulatory receptor as the protein stimulated by a fixation-sensitive ligand on the antigen-presenting cell (APC) displaying a stimulatory MHC-peptide complex8. Of note, few if any of these investigations were driven by “cancer research”.
This galaxy of investigation in turn led to families of co-stimulatory (4-1BB, ICOS, etc.) and co-inhibitory receptors (among which are CTLA-4 and PD-1, each a target of one class of pharmaceutical to enhance immune activity hitting tumors)9–11. In contrast to CD28, most of these crucial collaborators in TCR signal-interpretation are induced after T-cell activation. In parallel, the discovery of these regulators of T-cell activation, proliferation, and function prompted identification of their ligands, typically proteins expressed on the surfaces of APCs. These proteins that engage the co-stimulatory and -inhibitory receptors also follow a theme dividing constitutive (for instance, on so-called “professional APCs”) and inducible expression. Inflammation, including local cytokines but perhaps also intracellular sensors of cell stress (such as inflammasomes, Nod-like receptors, or cGAS), can promote induction of the ligands (e.g. PD-L1 or PD-L2) for the co-regulators (e.g. PD-1).
However, much of the emergence of effector functions for a naïve T cell takes place days after it first starts being activated, and intra-vital imaging reveals striking journeys traveled by the motile T lymphocyte and its daughters after an activating encounter12–15. At present, sufficient breadth and depth of information about these variable and less deterministic aspects of T-cell population development are sorely lacking, in part because of limitations in the present state of technology for such analyses. Accordingly, a missing link and frontier for investigation must be the time element and dynamism of influences on the T cell even after its initial encounter with an APC bearing stimulatory peptide-MHC complexes and cell-scale resolution of the variegated nutritional environments in which T cells operate.
Drilling in the message(s): TCR-activated signal transduction
The simple canonical outline of early signal transduction activated by TCR engagement and CD28 co-stimulation has been beautifully reviewed, most recently by Malissen16,17, who highlights points of uncertainty. In the general model, Src-family kinases initiate protein tyrosine phosphorylation at characteristic tyrosines in ITAMs (immunoreceptor tyrosine activation motifs) on the cytoplasmic tails of chains essential to the overall TCR complex. Phospho-ITAMs recruit adapter proteins crucial for signal propagation and diversification. One of these recruits is another protein tyrosine kinase, ZAP70, which phosphorylates yet further targets in the cascade. Of note, genetic and pharmacological analyses reveal ZAP70 as a prototype of a relay essential for one type of outcome from TCR engagement yet not another18–20. One adapter notable among the relays downstream from ZAP70, LAT, can interact with several distinct classes of further adapter and transducer proteins. This arrangement affords the capacity to diversify the nature of signal21–24. Elegant genetic experiments provide evidence of the essential nature of most of the proteins and even of distinct functions for specific tyrosines within their sequences21. As detailed in later sections, activity of multi-protein complexes containing the target of rapamycin (mTOR) serine-threonine kinase is induced downstream from these earliest and most rapid TCR-triggered signals.
Such monomorphic descriptions skip over vital parts of the body of data that probably are as crucial for the biology and the adaptive value of the system as its basic outline. First, let us consider the naïve T cell. Here, particular cases of individual TCR and their interactions with activating ligands indicated that quantitative and qualitative differences (the densities of peptide-MHC complexes and the exact sequence of the peptide) led not only to quantitative and qualitative differences in later-phase signaling (for instance, that of ERK MAP kinases) but also to altered effector fate and function25–29. A functional reinforcement of these fundamental findings emerged in elegant experiments seeking to sketch out the constraints on probabilities of an individual TCR yielding particular effector subsets in vivo by use of single-progenitor transfers30,31. In short, how a TCR signals varies according to the TCR. A second layer is that, even within the single-cell transfer results, the variance in distributions for a given TCR was substantial and probably reflects stochastic events that may even have a largely random origin.
So, an item for the future is to determine how much adaptive value in immunity builds on stochastic variation in the signaling from an individual TCR and the immune analogue of Heisenberg’s uncertainty principle. As a first step on this path, methods of single-cell analysis and modeling32–35 will be an exciting frontier. Older evidence showing that the diverse populations of memory T cells arising from a given TCR signal differently from their naïve progenitor population presents a third issue. In line with the points raised earlier in this paragraph, this picture is complicated by the fact that different results were obtained depending on the TCR. Finally, another exciting point is that the functional impact of a signal or its subcellular localization can depend on whether the TCR is on a suppressive, tumor-promoting regulatory T (Treg) cell or some other form of CD4 T cell36.
PI3K-mTOR signaling and the T cell
The lipid kinase phosphatidylinositol 3-kinase (PI3K) is another signal transduction mechanism initiated by the TCR or the combination of TCR and co-stimulatory receptor engagement. Notably, co-stimulatory receptors (e.g. CD28 and, even more strongly, ICOS) enhance generation of the lipid product of PI3K (i.e. phosphatidylinositol 3,4,5 triphosphate, or PIP3)37–42. This in turn enhances activity of serine-threonine kinases, including PIP3-dependent kinase-1 (PDK1) and its targets AKT, protein kinases C (PKC), and SGK1, as well as mTOR41–43 (Figure 1). Conversely, there is evidence that engagement of at least some of the co-inhibitory receptors decreases PI3K and mTOR activity44 but also that they stimulate these pathways45. Ultimately, these signals change gene expression profiles; alterations in DNA-binding transcription factors or other layers in the machinery for transcriptional regulation are probably a major part of the mechanism for such changes. Members of a branch of the Forkhead box transcription factor family (i.e. FoxO1 and FoxO3) are notable targets of these pathways. In particular, FoxO nuclear export and cytosolic retention are prompted by phosphorylation. The kinases for this negative regulation of FoxO are AKT – as regulated by phosphorylation by both PDK1 and mTOR in one of its two functional complexes – and SGK141,42,46. SGK1, like AKT, is activated by mTORC2. Though discussed less here because of constraints on length and focus, pertinent inputs from the stimulation of cytokine receptors by the complex mixtures of their ligands in the micro-environments of T cells also impact the behavior (survival, proliferation, migration, and differentiation) of lymphocytes. In aggregate, the sentences and paragraphs formed from the rich lexicon of cytokines are sometimes distilled to consideration of “signal 3” to integrate with the graded signals of TCR (signal 1) and co-stimulation/co-inhibition (signal 2).
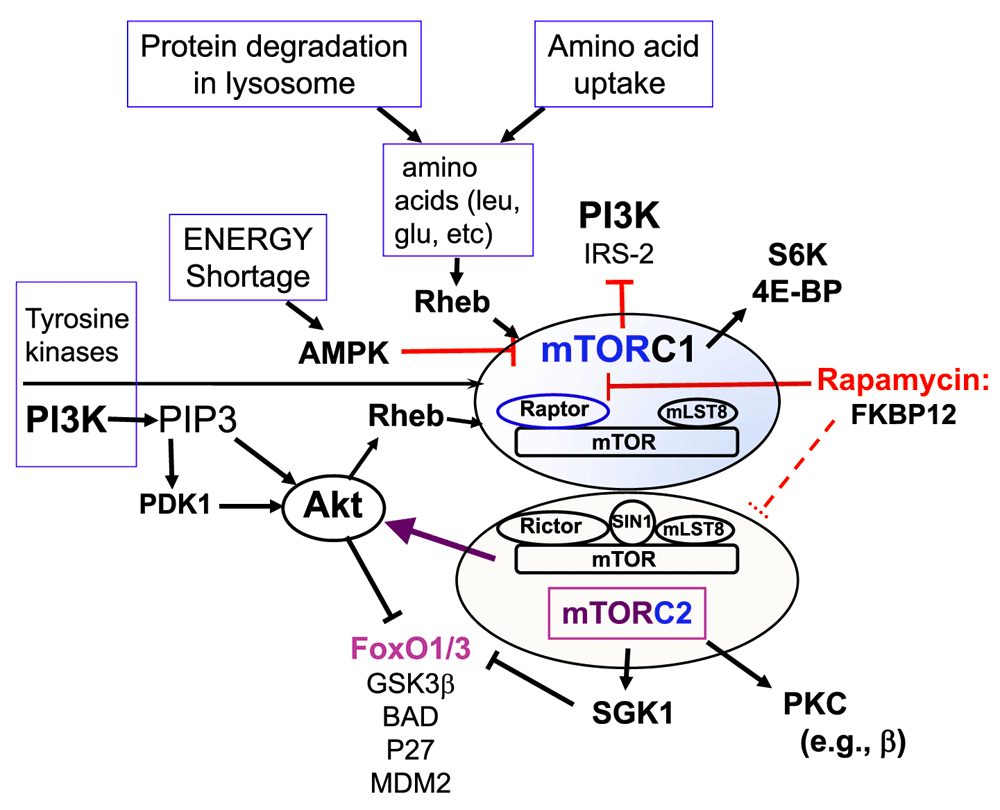
Figure 1. A simplified summary of the two distinct mammalian (or mechanistic) target of rapamycin (mTOR) signaling complexes, with some of the inputs (activators) and outputs.
Details are outlined in the text.
All together, this framework provides insights into – or “explains” – co-regulation, the various functional states of T cells, and the amount of functional activity these functional classes may retain (for instance, in the setting of anti-tumor responses of T cells). A central element of the story involves the capacity of CD4+ T cells to form various helper subsets but also both thymus-derived and peripherally generated suppressive Treg cells. Growth of a range of cancers is promoted (Treg) or restrained (cytotoxic CD8 T cells and some of the effector subsets) by subtypes of T cell47–50. Experiments pushing signaling to or beyond extremes of the dynamic range in physiology provided the result that persistently activated AKT blocked Treg fate51. These findings provide the crystal nucleus for a model in which modulation of the levels of AKT and mTOR provides a means to regulate a balance between taking on the Treg “fate” and that of T helper effectors such as the Th17 cell. Such a model harmonized with evidence derived from elimination of the enzyme mTOR from T lineage cells or inactivation of mTOR complex 252,53; in the extreme, this created a remarkable imbalance in which the effector fate after activation of naïve CD4+ T cells was almost completely diverted into that of cells expressing the “master regulator of Treg formation”, FoxP352.
In broader settings such as infection or cancer biology, of course, accounting for the involvement of and impact on thymus-derived Treg cells is a vital issue. Clearly, dominant suppression remains essential even after initial establishment of immune repertoires, since elimination of Treg from the mature mouse unleashed auto-inflammation54. Short of killing off FoxP3-expressing cells54, fate-marking experiments suggest that suppressive function is retained if FoxP3 is eliminated after establishment of the Treg state, although there is some divergence of findings and controversy in this area55,56. In a likely link to AKT, however, gene deletion analyses of the transcription factors FoxO1 and FoxO3a provide evidence that these transcription factors each regulate the formation of thymus-derived Treg cells but also that complete loss of FoxO1 undermined the functional capacity of FoxP3+ suppressors57–59. At one level, then, the collective findings yield a beautiful picture. Interference with PI3K-activating co-stimulation must decrease AKT-driven FoxO phosphorylation, thereby promoting its nuclear localization and Treg function.
Many further findings add paint to this model while also raising questions as to “who’s in charge of mTOR activity?” Most broadly, work of the past several years has increasingly highlighted that the capacity to supply amino acids to a juxta-lysosomal locale also appears crucial for activity of mTORC1 and pathways downstream from it60–63. Work with mouse systems provides evidence that G-protein-coupled receptors for complement fragments C3a and C5a are essential for Treg function and shows that most of the CD4 T cells’ mTOR activity is lost when both C3aR and C5aR are absent64,65. In parallel, a body of work with human CD4 T cells indicates that an accessory protein in the complement system, CD46, is particularly crucial for enhancing leucine uptake rates and mTORC1 activity a day after T-cell stimulation or co-stimulation by antibody cross-linking66. Though not tied definitively to FoxP3, the body of studies on complement and CD46 suggest that the suppressive activity yielded after activation is influenced through this pathway67. This work with human cells starts to get at one of the great gaps in the canon (i.e. the time element and an assessment of how mechanisms evolve within an individual T cell and its progeny as it moves through time and different locations). Thus, much of the evidence on signaling in T-cell activation focuses on time points that are quite early in relation to the time at which effector phenotype or an enhanced probability of memory fate arises. Work with mouse and human Treg cells emphasizes a vital role for the satiety-regulating hormone leptin in T-cell physiology along with a temporal function of leptin receptor-induced mTORC1 activity in repression of Treg proliferation68,69. This role of leptin in mTOR regulation, along with earlier evidence of its capacity to enhance interferon-gamma (IFN-γ) production from cultured CD4 T cells70, is intriguing in the context of metabolism of the patient with cancer. Another important aspect of mTOR regulation is suggested from the combination of recent and older work on signaling by Notch receptors. In addition to a requirement for Notch in thymic T-cell development, a variety of articles have linked this receptor to regulation of the balance of effector cell subset formation, including evidence for Notch-stimulated mTOR controlling Treg71–82. Of note, Notch stimulates both mTORC1 and mTORC2 activity; each of these branches is important for T helper subset specification, including distinct functions of mTORC1 and mTORC2 in Treg53,82–85, and is needed for Notch function in thymocytes84,85. PI3K-mTOR activity initiated by the inducible co-stimulator ICOS is yet another driver of mature T-cell differentiation (e.g. the follicular helper subset, TFH)40,86.
So, what could be the m(a)TOR with the beautiful picture?
Notwithstanding the beautiful picture, the PI3K-mTOR signaling pathway further exemplifies some pitfalls of relying on a binary style of drawing conclusions and the previously noted complications in conceiving of T-cell differentiation independent from the dynamism of time and space (hence, micro-environment). At present, the dominant experimental tool consists of stable, nearly complete losses of function. This suggests a need for caution about physiological perturbation-response relationships (Figure 2). This concern is strengthened by evidence that the magnitude and duration of mTOR kinase activation vary according to the circumstances of stimulation (including the mix and timing of cytokine exposures after initial activation) and work showing that the effects of rapamycin depend on the strength and nature of stimulation87,88. The preceding section suggests yet another gap in our approaches and understanding. As ever more phenomena are attributed to the activation of mTOR or its nearly complete failure — e.g. 89,90 — in addition to previously cited work (e.g. 39,40,61,62,65,66,69), it will be intriguing to sort out mechanisms; already, well over a half-dozen disparate receptors with very different ligands are each reportedly necessary for approximately 80% (or more) of the activity on a branch of the mTOR pathway.
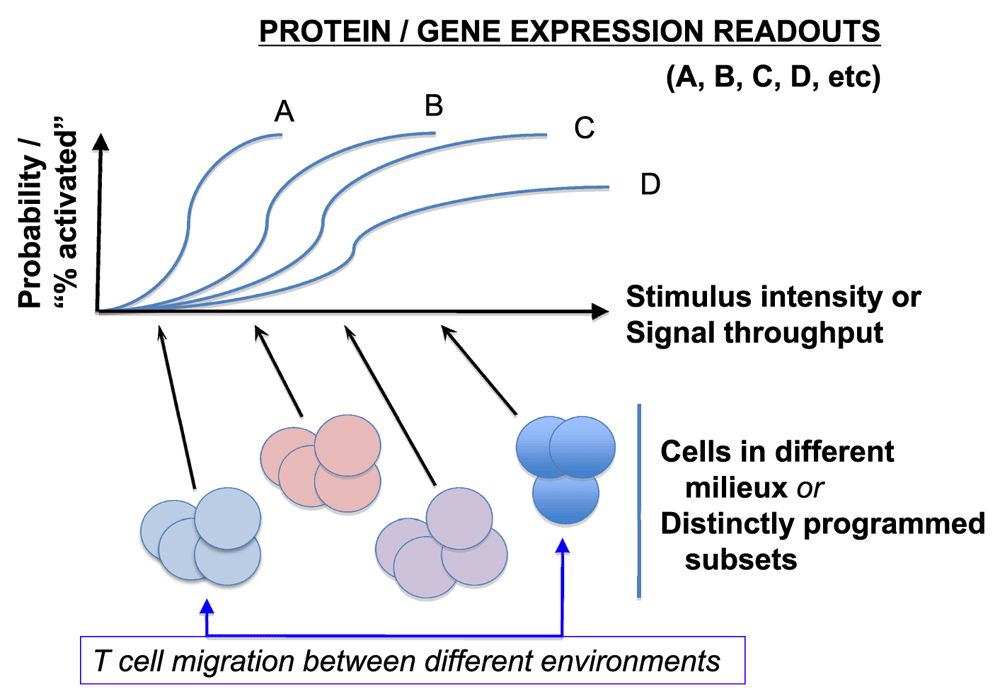
Figure 2. Challenges in extrapolating complete (or almost complete) and permanent loss-of-function results to sensors poised with different dose-response curves and threshold settings in biological ranges.
As highlighted by evolving single-cell stimulus-response research34,35, a given stimulus will often yield a probability of evoked response well below 1, and this stimulus-probability curve will vary within a series of gene expression or downstream signal intensities pertinent to T-cell differentiation or function. Accordingly, as cell conditions or pre-existing programs vary or as different extents of impairment to a given signal relay are imposed, the responses will exhibit variegation among cells in a population. Similar principles are postulated almost certainly to apply to the time element (e.g. how long does it take for a condition to change histone post-translational modifications or epigenetic modification of DNA or to change the level of expression of a target gene?). As noted in Figure 4, local conditions vary even within a single tumor mass (setting aside known differences between metastatic and primary tumors).
There might be some super-complex requiring all these disparate elements at once. Also, there may prove to be subtle variations in the time course or protocols even as the nature of developing scientific stories emphasizes “optimizing conditions” to maximize a particular observation. On a mechanistic note, one potential resolution of the conundrum is whether there are relay or cross-talk effects (e.g. whether mTOR activity stimulated by one receptor depends on antecedent induction by a different stimulus). This model may apply in the case of Notch-stimulated mTOR, which then feeds into “tuning” TCR sensitivity73, a model separately proposed for the transcription factor nuclear factor-kappa-B (NF-κB) in thymocytes91. Another possibility links to changes in subcellular localization. Key kinases in this pathway partition among compartments (for instance, AKT and the mTORC1 target S6K)92–95. In the case of FoxO phosphorylation, it is appealing to consider that nuclear AKT is a crucial element of the equation. Of note, the efficiency of nuclear sampling for phosphorylated forms of AKT varies dramatically among means of cell extraction (an analytic problem that is even worse with intracellular staining for flow cytometry). So, there may be differential sampling of these compartments (e.g. nucleus versus cytosol) in various articles. Among the mutually compatible possibilities is that function is effected in large part via relays and supra-molecular complexes akin to signaling involving MAPK or NF-κB activation96–99.
Second, important work on FoxO1 and stability of immune homeostasis mediated by Treg used a knock-in that permitted quantitation of the extent to which TCR stimulation could evict Foxo1 from the nucleus. In this analysis, two key findings were that (i) for FoxP3+ (Treg) and FoxP3− (Tconv) cells, signal intensities for both Erk and the mTOR–AKT–FoxO1 were quite different, as was the extent of FoxO1 redistribution, and (ii) at best, partial nuclear exclusion was driven when focusing on Treg59. Accordingly, even haplo-insufficiency for FoxO1 may overestimate the extreme end of a dynamic range that is achieved in vivo. An intriguing side point pertaining to transforming growth factor-beta-rich tumor environments is that this cytokine activates mTOR and AKT100–102 even though AKT is supposed to suppress Treg fate or function. In light of the evidence that signal distribution and “interpretation” within Treg can differ dramatically from those in the conventional CD4+ T cell, detailed analysis of how PD-1 actually affects mTORC1, mTORC2, and other signaling pathways is needed. More broadly, there is a need in the field to seem less absolute and oversimplistic. In addition to the factors already noted, recent work in a developmental system provides strong evidence that the use of longer-term and complete gene inactivation or loss of function can elicit compensatory circuits that are avoided when a signal is attenuated more acutely with RNA interference (RNAi)103. As one working example, disparate articles even within the spheres of both CTL and conventional helper T-cell differentiation observe quite different outcomes (both qualitatively and quantitatively) with perturbing the same pathway53,79–81,83,104–107 (Figure 3). These differences probably represent combinations of fine gradations in the timing and extent of loss-of-function perturbations and hence distinct points on uncharacterized dose-response curves, along with variance among models and, who knows, maybe even microbiomes.
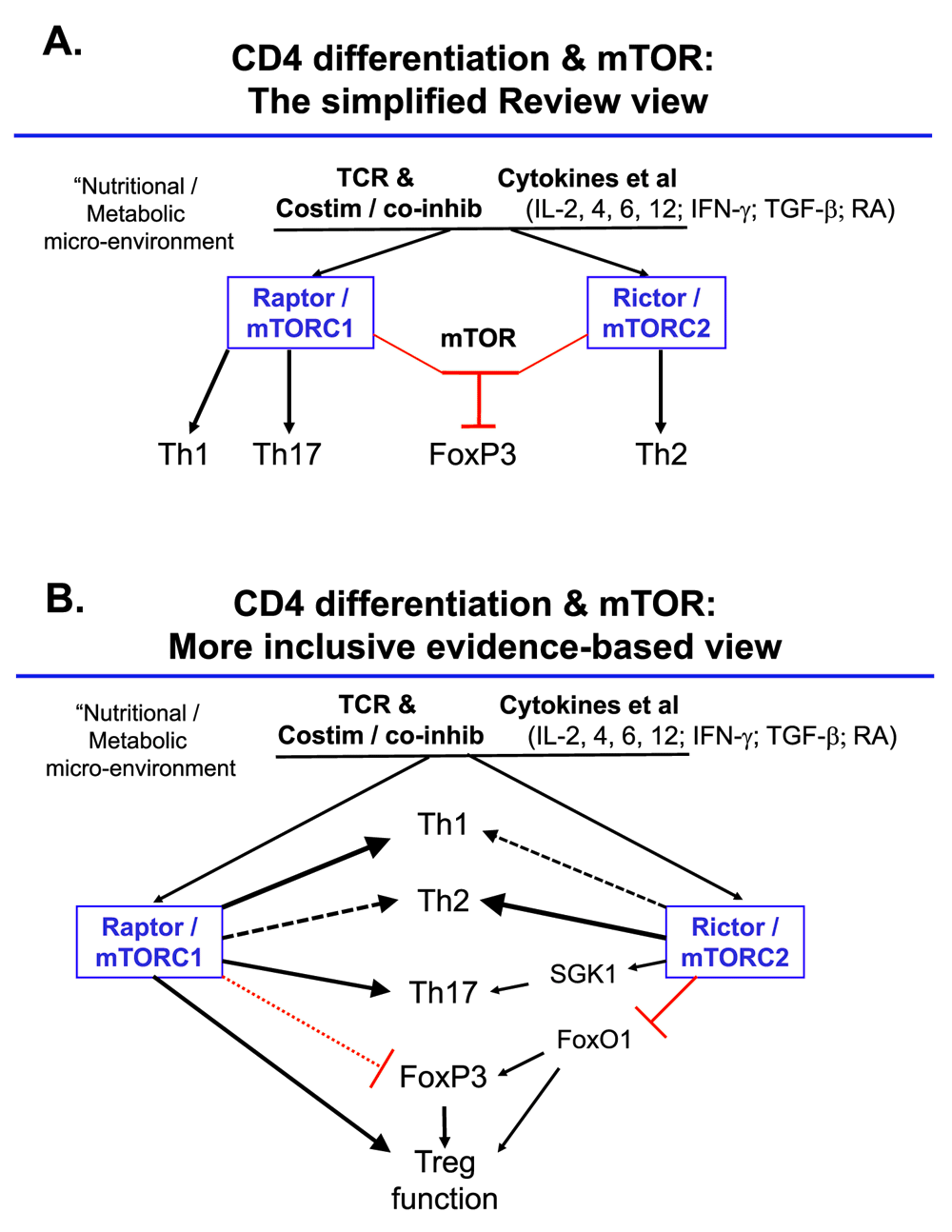
Figure 3. Over-simplified and more literature-based pictures of mTOR regulation of the acquired functions of mature CD4+ T-cells.
(A) An oversimplified view segregating functions of mammalian (or mechanistic) target of rapamycin (mTOR) complexes with distinct functions mapping one on one with T helper (Th) subsets. (B) A partial integration of more complete information as represented in the cited articles51–53,57–59,79–83 and related work. Notable differences include that, presumably as a function of differences in conditions, the literature supports roles for each mTOR complex in promoting each of the peripherally acquired CD4 effector phenotypes (Th1, Th2, Th17), while the effects on regulatory T (Treg) are complex, with mTOR and mTORC2 shown to restrain induction of FoxP352,53 but mTORC1 vital for the suppressive function of these cells83. If non-drastic decreases in nuclear FoxO1 are assumed to attenuate Treg function59, then enhanced mTORC2 activity in Treg might decrease their inhibitory properties. The connection of mTORC2 to Th17 function is inferred from mTORC2 activation of SGK1 and the function of this latter factor in promoting pathogenic Th1746. Abbreviations: IFN-γ, interferon-gamma; IL, interleukin; RA, retinoic acid; TCR, T-cell receptor; TGF-β, transforming growth factor-beta.
Third, T cells are quite the moving target. Intra-vital imaging, albeit with caveats relating to the potential impact of clonal frequency on responses108,109, shows that after a period of arrest, T cells resume roaming within the lymphoid organ. Moreover, activated T cells leave the lymphoid organs to circulate and be recruited to tissues – especially if there are sites of inflammation. Further complexities related to inflammation and the kaleidoscope of the micro-environment include hypoxia, angiogenesis, and tumor-associated myeloid-lineage cells. The metabolic landscape differs among the arterial, venous, and lymphatic circulations as well as the tissues, which in turn will be different if challenged by metastatic cancer cells or microbial invasion (e.g. intracellular pathogens) (Figure 4). Co-stimulation and hypoxia response mechanisms impact motility, mTOR activities, and T-cell phenotypes, although sometimes with opposite experimental results that may depend on the metabolic environment of the tested cells, the lymphocyte subset, whether the T cell had previously been activated, and the form of stimulus (e.g. cytokine)110–115. As a tumor grows, mutability can yield “self” peptides of altered sequence that may then be perceived as foreign (“non-self”)116. Indeed, new evidence suggests a correlation or even mechanistic connection between the degree to which such epitopes are generated and the responsiveness to checkpoint inhibitors117,118. However, the universe of peptides that activate Treg may differ from those that engage TCR on conventional CD4 and CD8 T cells, and MHC-neopeptide complexes will include antagonists that drive clonal anergy119.
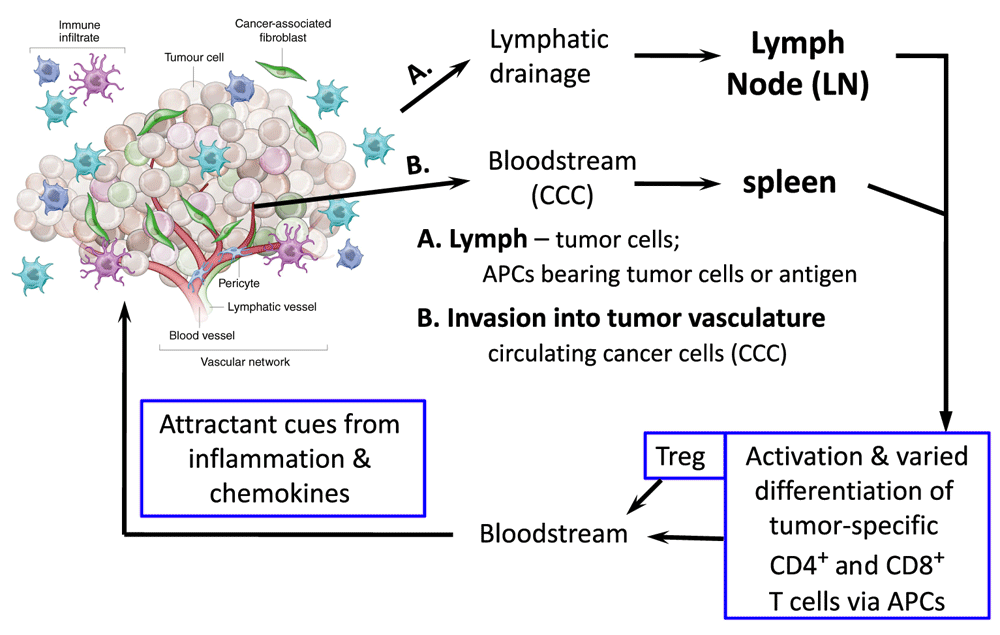
Figure 4. T cells and the tumor.
Shown is a simplified overview of the multiple and diverse sites at which each type of T cell may encounter signals and tumor-derived antigens during growth and metastasis for carcinomas, together with a diagram underscoring likely variations of malignant cells within the tumor mass itself. In addition to action within the tumor itself, which may not be a site of initial tumor antigen presentation to and activation of T cells, either live or dead tumor cells, or antigens derived from them, traffic and flow to secondary lymphoid organs (spleen; lymph node), in either particulate or cell-associated (e.g. within a dendritic or movable phagocytic cell) form. APC, antigen-presenting cell; Treg, regulatory T.
Imagining parts of a future
To recap, an attractive model based on “PI3K–AKT–mTOR” signals that drastically alter nuclear FoxO protein levels could link checkpoint therapies to changes in immune responsiveness controlled by Treg. However, constrained by the state of current technologies, the first-wave studies are based on relatively extreme loss- or gain-of-function changes that are fixed and static. In the worst instances, inferences are drawn and conclusions are strongly stated based on knockouts that yield starting T-cell populations identifiably different from the reference “wild-type controls”. But even setting aside this problem, temperance and caution are called for in making a dogma from reasonable inferences based on immutable perturbations in systems that have clear feedback inhibition and counter-regulation. Moreover, evidence is lacking as to where key changes in the relevant cells occur – with the cells in question highly dynamic. It is likely a priori that shifts in the environment as activated T cells move within and out from tissues modulate the probabilities of particular fates (Figure 4).
Looking ahead, then, it is suggested that the list of needs includes the following:
1. Cultural change in science, with fewer oversimplifications that rely on overly categorical divisions between function of A versus function of B (Figure 3). A central challenge stems from a combination of the overly definitive presentation of findings along with an implicit expectation of relatively universal “explanations” and an undue degree of “buy-in” to static conceptual schemata (explanatory cartoons).
2. More information on heterozygote phenotypes for loss-of-function perturbations of these pathways rather than focusing exclusively on the extremes – more broadly, better and more widespread attention to and precision with dose-response curves and understanding of stochastic variance (Figure 2).
3. Development and use of “tools” and methodologies whereby graded changes in activity of the systems can be imposed – ideally, for more limited periods of time even than a laudable model making use of 4-OH-tamoxifen-activated estrogen receptor ligand-binding domains120. Application of optogenetic tools or their marriage to new genetics may prove valuable in moving toward this goal, ideally with the added value of permitting spatially restricted activation121–124.
4. More ready supply of existing tools, along with development of better means for analyzing the actual pool of fuels (e.g. hexoses, glutamine, fatty acids, and oxygen) and metabolites in the interstitial spaces which are the soils among which cells are moving, along with
5. A capacity to follow the cells and better detect their signaling in situ124–126 across the timescales relevant to their development and functions.
Competing interests
The author declares that he has no competing interests.
Grant information
The author(s) declared that no grants were involved in supporting this work.
Faculty Opinions recommendedReferences
- 1.
Couzin-Frankel J:
Breakthrough of the year 2013. Cancer immunotherapy.
Science.
2013; 342(6165): 1432–3. PubMed Abstract
| Publisher Full Text
- 2.
Topalian SL, Hodi FS, Brahmer JR, et al.:
Safety, activity, and immune correlates of anti-PD-1 antibody in cancer.
N Engl J Med.
2012; 366(26): 2443–54. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 3.
Brahmer JR, Tykodi SS, Chow LQ, et al.:
Safety and activity of anti-PD-L1 antibody in patients with advanced cancer.
N Engl J Med.
2012; 366(26): 2455–65. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 4.
Bretscher P, Cohn M:
A theory of self-nonself discrimination.
Science.
1970; 169(3950): 1042–9. PubMed Abstract
| Publisher Full Text
- 5.
Matis LA:
The molecular basis of T-cell specificity.
Annu Rev Immunol.
1990; 8: 65–82. PubMed Abstract
| Publisher Full Text
- 6.
Garcia KC:
Molecular interactions between extracellular components of the T-cell receptor signaling complex.
Immunol Rev.
1999; 172(1): 73–85. PubMed Abstract
| Publisher Full Text
- 7.
Jenkins MK, Schwartz RH:
Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo.
J Exp Med.
1987; 165(2): 302–19. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 8.
Harding FA, McArthur JG, Gross JA, et al.:
CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones.
Nature.
1992; 356(6370): 607–9. PubMed Abstract
| Publisher Full Text
- 9.
Freeman GJ, Long AJ, Iwai Y, et al.:
Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation.
J Exp Med.
2000; 192(7): 1027–34. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 10.
Nishimura H, Okazaki T, Tanaka Y, et al.:
Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice.
Science.
2001; 291(5502): 319–22. PubMed Abstract
| Publisher Full Text
- 11.
Greenwald RJ, Freeman GJ, Sharpe AH:
The B7 family revisited.
Annu Rev Immunol.
2005; 23: 515–48. PubMed Abstract
| Publisher Full Text
- 12.
Bajénoff M, Egen JG, Koo LY, et al.:
Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes.
Immunity.
2006; 25(6): 989–1001. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 13.
Engelhardt JJ, Boldajipour B, Beemiller P, et al.:
Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells.
Cancer Cell.
2012; 21(3): 402–17. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 14.
Shulman Z, Gitlin AD, Targ S, et al.:
T follicular helper cell dynamics in germinal centers.
Science.
2013; 341(6146): 673–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 15.
Honda T, Egen JG, Lämmermann T, et al.:
Tuning of antigen sensitivity by T cell receptor-dependent negative feedback controls T cell effector function in inflamed tissues.
Immunity.
2014; 40(2): 235–47. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 16.
Smith-Garvin JE, Koretzky GA, Jordan MS:
T cell activation.
Annu Rev Immunol.
2009; 27: 591–619. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 17.
Malissen B, Bongrand P:
Early T cell activation: integrating biochemical, structural, and biophysical cues.
Annu Rev Immunol.
2015; 33: 539–61. PubMed Abstract
| Publisher Full Text
- 18.
Au-Yeung BB, Levin SE, Zhang C, et al.:
A genetically selective inhibitor demonstrates a function for the kinase Zap70 in regulatory T cells independent of its catalytic activity.
Nat Immunol.
2010; 11(12): 1085–92. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 19.
Au-Yeung BB, Melichar HJ, Ross JO, et al.:
Quantitative and temporal requirements revealed for Zap70 catalytic activity during T cell development.
Nat Immunol.
2014; 15(7): 687–94. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 20.
Klammt C, Novotná L, Li DT, et al.:
T cell receptor dwell times control the kinase activity of Zap70.
Nat Immunol.
2015; 16(9): 961–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 21.
Aguado E, Richelme S, Nuñez-Cruz S, et al.:
Induction of T helper type 2 immunity by a point mutation in the LAT adaptor.
Science.
2002; 296(5575): 2036–40. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 22.
Samelson LE:
Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins.
Annu Rev Immunol.
2002; 20: 371–94. PubMed Abstract
| Publisher Full Text
- 23.
Balagopalan L, Coussens NP, Sherman E, et al.:
The LAT story: a tale of cooperativity, coordination, and choreography.
Cold Spring Harb Perspect Biol.
2010; 2(8): a005512. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 24.
Bezman N, Koretzky GA:
Compartmentalization of ITAM and integrin signaling by adapter molecules.
Immunol Rev.
2007; 218(1): 9–28. PubMed Abstract
| Publisher Full Text
- 25.
Evavold BD, Allen PM:
Separation of IL-4 production from Th cell proliferation by an altered T cell receptor ligand.
Science.
1991; 252(5010): 1308–10. PubMed Abstract
| Publisher Full Text
- 26.
Constant SL, Bottomly K:
Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches.
Annu Rev Immunol.
1997; 15: 297–322. PubMed Abstract
| Publisher Full Text
- 27.
Tao X, Constant S, Jorritsma P, et al.:
Strength of TCR signal determines the costimulatory requirements for Th1 and Th2 CD4+ T cell differentiation.
J Immunol.
1997; 159(12): 5956–63. PubMed Abstract
- 28.
Chang CF, D'Souza WN, Ch'en IL, et al.:
Polar opposites: Erk direction of CD4 T cell subsets.
J Immunol.
2012; 189(2): 721–31. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 29.
Mingueneau M, Krishnaswamy S, Spitzer MH, et al.:
Single-cell mass cytometry of TCR signaling: amplification of small initial differences results in low ERK activation in NOD mice.
Proc Natl Acad Sci U S A.
2014; 111(46): 16466–71. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 30.
Tubo NJ, Pagán AJ, Taylor JJ, et al.:
Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection.
Cell.
2013; 153(4): 785–96. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 31.
Nelson RW, Rajpal MN, Jenkins MK:
The Neonatal CD4+ T Cell Response to a Single Epitope Varies in Genetically Identical Mice.
J Immunol.
2015; 195(5): 2115–21. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 32.
Brennecke P, Reyes A, Pinto S, et al.:
Single-cell transcriptome analysis reveals coordinated ectopic gene-expression patterns in medullary thymic epithelial cells.
Nat Immunol.
2015; 16(9): 933–41. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 33.
Meredith M, Zemmour D, Mathis D, et al.:
Aire controls gene expression in the thymic epithelium with ordered stochasticity.
Nat Immunol.
2015; 16(9): 942–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 34.
Neuert G, Munsky B, Tan RZ, et al.:
Systematic identification of signal-activated stochastic gene regulation.
Science.
2013; 339(6119): 584–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 35.
Fang M, Xie H, Dougan SK, et al.:
Stochastic cytokine expression induces mixed T helper cell States.
PLoS Biol.
2013; 11(7): e1001618. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 36.
Zanin-Zhorov A, Ding Y, Kumari S, et al.:
Protein kinase C-theta mediates negative feedback on regulatory T cell function.
Science.
2010; 328(5976): 372–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 37.
Truitt KE, Shi J, Gibson S, et al.:
CD28 delivers costimulatory signals independently of its association with phosphatidylinositol 3-kinase.
J Immunol.
1995; 155(10): 4702–10. PubMed Abstract
- 38.
Hutchcroft JE, Bierer BE:
Signaling through CD28/CTLA-4 family receptors: puzzling participation of phosphatidylinositol-3 kinase.
J Immunol.
1996; 156(11): 4071–4. PubMed Abstract
- 39.
Dodson LF, Boomer JS, Deppong CM, et al.:
Targeted knock-in mice expressing mutations of CD28 reveal an essential pathway for costimulation.
Mol Cell Biol.
2009; 29(13): 3710–21. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 40.
Gigoux M, Shang J, Pak Y, et al.:
Inducible costimulator promotes helper T-cell differentiation through phosphoinositide 3-kinase.
Proc Natl Acad Sci U S A.
2009; 106(48): 20371–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 41.
Fingar DC, Blenis J:
Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression.
Oncogene.
2004; 23(18): 3151–71. PubMed Abstract
| Publisher Full Text
- 42.
Laplante M, Sabatini DM:
mTOR signaling in growth control and disease.
Cell.
2012; 149(2): 274–93. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 43.
Liu P, Gan W, Chin YR, et al.:
PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex.
Cancer Discov.
2015; 5(11): 1194–209. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 44.
Parry RV, Chemnitz JM, Frauwirth KA, et al.:
CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms.
Mol Cell Biol.
2005; 25(21): 9543–53. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 45.
Schneider H, Valk E, Leung R, et al.:
CTLA-4 activation of phosphatidylinositol 3-kinase (PI 3-K) and protein kinase B (PKB/AKT) sustains T-cell anergy without cell death.
PLoS One.
2008; 3(12): e3842. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 46.
Wu C, Yosef N, Thalhamer T, et al.:
Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1.
Nature.
2013; 496(7446): 513–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 47.
Sutmuller RP, van Duivenvoorde LM, van Elsas A, et al.:
Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25+ regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses.
J Exp Med.
2001; 194(6): 823–32. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 48.
Van Meirvenne S, Dullaers M, Heirman C, et al.:
In vivo depletion of CD4+CD25+ regulatory T cells enhances the antigen-specific primary and memory CTL response elicited by mature mRNA-electroporated dendritic cells.
Mol Ther.
2005; 12(5): 922–32. PubMed Abstract
| Publisher Full Text
- 49.
Fourcade J, Sun Z, Pagliano O, et al.:
PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8+ T cells induced by melanoma vaccines.
Cancer Res.
2014; 74(4): 1045–55. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 50.
Karimi S, Chattopadhyay S, Chakraborty NG:
Manipulation of regulatory T cells and antigen-specific cytotoxic T lymphocyte-based tumour immunotherapy.
Immunology.
2015; 144(2): 186–96. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 51.
Haxhinasto S, Mathis D, Benoist C:
The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells.
J Exp Med.
2008; 205(3): 565–74. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 52.
Delgoffe GM, Kole TP, Zheng Y, et al.:
The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment.
Immunity.
2009; 30(6): 832–44. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 53.
Lee K, Gudapati P, Dragovic S, et al.:
Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways.
Immunity.
2010; 32(6): 743–53. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 54.
Kim JM, Rasmussen JP, Rudensky AY:
Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice.
Nat Immunol.
2007; 8(2): 191–7. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 55.
Rubtsov YP, Niec RE, Josefowicz S, et al.:
Stability of the regulatory T cell lineage in vivo.
Science.
2010; 329(5999): 1667–71. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 56.
Sakaguchi S, Vignali DA, Rudensky AY, et al.:
The plasticity and stability of regulatory T cells.
Nat Rev Immunol.
2013; 13(6): 461–7. PubMed Abstract
| Publisher Full Text
- 57.
Ouyang W, Beckett O, Ma Q, et al.:
Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells.
Nat Immunol.
2010; 11(7): 618–27. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 58.
Kerdiles YM, Stone EL, Beisner DR, et al.:
Foxo transcription factors control regulatory T cell development and function.
Immunity.
2010; 33(6): 890–904. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 59.
Ouyang W, Liao W, Luo CT, et al.:
Novel Foxo1-dependent transcriptional programs control Treg cell function.
Nature.
2012; 491(7425): 554–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 60.
Bar-Peled L, Sabatini DM:
Regulation of mTORC1 by amino acids.
Trends Cell Biol.
2014; 24(7): 400–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 61.
Sinclair LV, Rolf J, Emslie E, et al.:
Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation.
Nat Immunol.
2013; 14(5): 500–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 62.
Nakaya M, Xiao Y, Zhou X, et al.:
Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation.
Immunity.
2014; 40(5): 692–705. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 63.
Klysz D, Tai X, Robert PA, et al.:
Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation.
Sci Signal.
2015; 8(396): ra97. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 64.
Strainic MG, Liu J, Huang D, et al.:
Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells.
Immunity.
2008; 28(3): 425–35. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 65.
Kwan W, van der Touw W, Paz-Artal E, et al.:
Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells.
J Exp Med.
2013; 210(2): 257–68. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 66.
Kolev M, Dimeloe S, Le Friec G, et al.:
Complement Regulates Nutrient Influx and Metabolic Reprogramming during Th1 Cell Responses.
Immunity.
2015; 42(6): 1033–47. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 67.
Barchet W, Price JD, Cella M, et al.:
Complement-induced regulatory T cells suppress T-cell responses but allow for dendritic-cell maturation.
Blood.
2006; 107(4): 1497–504. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 68.
Procaccini C, De Rosa V, Galgani M, et al.:
An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness.
Immunity.
2010; 33(6): 929–41. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 69.
Procaccini C, De Rosa V, Galgani M, et al.:
Leptin-induced mTOR activation defines a specific molecular and transcriptional signature controlling CD4+ effector T cell responses.
J Immunol.
2012; 189(6): 2941–53. PubMed Abstract
| Publisher Full Text
- 70.
Lord GM, Matarese G, Howard JK, et al.:
Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression.
Nature.
1998; 394(6696): 897–901. PubMed Abstract
| Publisher Full Text
- 71.
Minter LM, Turley DM, Das P, et al.:
Inhibitors of gamma-secretase block in vivo and in vitro T helper type 1 polarization by preventing Notch upregulation of Tbx21.
Nat Immunol.
2005; 6(7): 680–8. PubMed Abstract
| Publisher Full Text
- 72.
Eagar TN, Tang Q, Wolfe M, et al.:
Notch 1 signaling regulates peripheral T cell activation.
Immunity.
2004; 20(4): 407–15. PubMed Abstract
| Publisher Full Text
- 73.
Laky K, Evans S, Perez-Diez A, et al.:
Notch signaling regulates antigen sensitivity of naive CD4+ T cells by tuning co-stimulation.
Immunity.
2015; 42(1): 80–94. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 74.
Radtke F, Wilson A, Stark G, et al.:
Deficient T cell fate specification in mice with an induced inactivation of Notch1.
Immunity.
1999; 10(5): 547–58. PubMed Abstract
| Publisher Full Text
- 75.
Tanigaki K, Tsuji M, Yamamoto N, et al.:
Regulation of alphabeta/gammadelta T cell lineage commitment and peripheral T cell responses by Notch/RBP-J signaling.
Immunity.
2004; 20(5): 611–22. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 76.
Amsen D, Antov A, Jankovic D, et al.:
Direct regulation of Gata3 expression determines the T helper differentiation potential of Notch.
Immunity.
2007; 27(1): 89–99. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 77.
Amsen D, Antov A, Flavell RA:
The different faces of Notch in T-helper-cell differentiation.
Nat Rev Immunol.
2009; 9(2): 116–24. PubMed Abstract
| Publisher Full Text
- 78.
Dongre A, Surampudi L, Lawlor RG, et al.:
Non-Canonical Notch Signaling Drives Activation and Differentiation of Peripheral CD4+ T Cells.
Front Immunol.
2014; 5: 54. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 79.
Delgoffe GM, Pollizzi KN, Waickman AT, et al.:
The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2.
Nat Immunol.
2011; 12(4): 295–303. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 80.
Kurebayashi Y, Nagai S, Ikejiri A, et al.:
PI3K-Akt-mTORC1-S6K1/2 axis controls Th17 differentiation by regulating Gfi1 expression and nuclear translocation of RORγ.
Cell Rep.
2012; 1(4): 360–73. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 81.
Yang K, Shrestha S, Zeng H, et al.:
T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming.
Immunity.
2013; 39(6): 1043–56. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 82.
Kared H, Adle-Biassette H, Foïs E, et al.:
Jagged2-expressing hematopoietic progenitors promote regulatory T cell expansion in the periphery through notch signaling.
Immunity.
2006; 25(5): 823–34. PubMed Abstract
| Publisher Full Text
- 83.
Zeng H, Yang K, Cloer C, et al.:
mTORC1 couples immune signals and metabolic programming to establish Treg-cell function.
Nature.
2013; 499(7459): 485–90. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 84.
Lee K, Nam KT, Cho SH, et al.:
Vital roles of mTOR complex 2 in Notch-driven thymocyte differentiation and leukemia.
J Exp Med.
2012; 209(4): 713–28. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 85.
Wong GW, Knowles GC, Mak TW, et al.:
HES1 opposes a PTEN-dependent check on survival, differentiation, and proliferation of TCRβ-selected mouse thymocytes.
Blood.
2012; 120(7): 1439–48. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 86.
Stone EL, Pepper M, Katayama CD, et al.:
ICOS coreceptor signaling inactivates the transcription factor FOXO1 to promote Tfh cell differentiation.
Immunity.
2015; 42(2): 239–51. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 87.
Slavik JM, Lim DG, Burakoff SJ, et al.:
Uncoupling p70s6 kinase activation and proliferation: rapamycin-resistant proliferation of human CD8+ T lymphocytes.
J Immunol.
2001; 166(5): 3201–9. PubMed Abstract
| Publisher Full Text
- 88.
Li Q, Rao R, Vazzana J, et al.:
Regulating mammalian target of rapamycin to tune vaccination-induced CD8+ T cell responses for tumor immunity.
J Immunol.
2012; 188(7): 3080–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 89.
Chang J, Burkett PR, Borges CM, et al.:
MyD88 is essential to sustain mTOR activation necessary to promote T helper 17 cell proliferation by linking IL-1 and IL-23 signaling.
Proc Natl Acad Sci U S A.
2013; 110(6): 2270–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 90.
Delgoffe GM, Woo SR, Turnis ME, et al.:
Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis.
Nature.
2013; 501(7466): 252–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 91.
Mora AL, Stanley S, Armistead W, et al.:
Inefficient ZAP-70 phosphorylation and decreased thymic selection in vivo result from inhibition of NF-kappaB/Rel.
J Immunol.
2001; 167(10): 5628–35. PubMed Abstract
| Publisher Full Text
- 92.
Astoul E, Watton S, Cantrell D:
The dynamics of protein kinase B regulation during B cell antigen receptor engagement.
J Cell Biol.
1999; 145(7): 1511–20. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 93.
Panasyuk G, Nemazanyy I, Zhyvoloup A, et al.:
Nuclear export of S6K1 II is regulated by protein kinase CK2 phosphorylation at Ser-17.
J Biol Chem.
2006; 281(42): 31188–201. PubMed Abstract
| Publisher Full Text
- 94.
Liu JL, Mao Z, LaFortune TA, et al.:
Cell cycle-dependent nuclear export of phosphatase and tensin homologue tumor suppressor is regulated by the phosphoinositide-3-kinase signaling cascade.
Cancer Res.
2007; 67(22): 11054–63. PubMed Abstract
| Publisher Full Text
- 95.
Rosner M, Hengstschläger M:
Nucleocytoplasmic localization of p70 S6K1, but not of its isoforms p85 and p31, is regulated by TSC2/mTOR.
Oncogene.
2011; 30(44): 4509–22. PubMed Abstract
| Publisher Full Text
- 96.
Morrison DK, Davis RJ:
Regulation of MAP kinase signaling modules by scaffold proteins in mammals.
Annu Rev Cell Dev Biol.
2003; 19: 91–118. PubMed Abstract
| Publisher Full Text
- 97.
Wortzel I, Seger R:
The ERK Cascade: Distinct Functions within Various Subcellular Organelles.
Genes Cancer.
2011; 2(3): 195–209. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 98.
Zhang SQ, Kovalenko A, Cantarella G, et al.:
Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation.
Immunity.
2000; 12(3): 301–11. PubMed Abstract
| Publisher Full Text
- 99.
Paul S, Traver MK, Kashyap AK, et al.:
T cell receptor signals to NF-κB are transmitted by a cytosolic p62-Bcl10-Malt1-IKK signalosome.
Sci Signal.
2014; 7(325): ra45. PubMed Abstract
| Publisher Full Text
- 100.
Bakin AV, Tomlinson AK, Bhowmick NA, et al.:
Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration.
J Biol Chem.
2000; 275(47): 36803–10. PubMed Abstract
| Publisher Full Text
- 101.
Das F, Bera A, Ghosh-Choudhury N, et al.:
TGFβ-induced deptor suppression recruits mTORC1 and not mTORC2 to enhance collagen I (α2) gene expression.
PLoS One.
2014; 9(10): e109608. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 102.
Abe Y, Sakairi T, Beeson C, et al.:
TGF-β1 stimulates mitochondrial oxidative phosphorylation and generation of reactive oxygen species in cultured mouse podocytes, mediated in part by the mTOR pathway.
Am J Physiol Renal Physiol.
2013; 305(10): F1477–90. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 103.
Rossi A, Kontarakis Z, Gerri C, et al.:
Genetic compensation induced by deleterious mutations but not gene knockdowns.
Nature.
2015; 524(7564): 230–3. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 104.
Pearce EL, Walsh MC, Cejas PJ, et al.:
Enhancing CD8 T-cell memory by modulating fatty acid metabolism.
Nature.
2009; 460(7251): 103–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 105.
Araki K, Turner AP, Shaffer VO, et al.:
mTOR regulates memory CD8 T-cell differentiation.
Nature.
2009; 460(7251): 108–12. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 106.
Li Q, Rao RR, Araki K, et al.:
A central role for mTOR kinase in homeostatic proliferation induced CD8+ T cell memory and tumor immunity.
Immunity.
2011; 34(4): 541–53. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 107.
Pollizzi KN, Patel CH, Sun IH, et al.:
mTORC1 and mTORC2 selectively regulate CD8+ T cell differentiation.
J Clin Invest.
2015; 125(5): 2090–108. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 108.
Badovinac VP, Haring JS, Harty JT:
Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8+ T cell response to infection.
Immunity.
2007; 26(6): 827–41. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 109.
Wirth TC, Harty JT, Badovinac VP:
Modulating numbers and phenotype of CD8+ T cells in secondary immune responses.
Eur J Immunol.
2010; 40(7): 1916–26. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 110.
Thauland TJ, Koguchi Y, Dustin ML, et al.:
CD28-CD80 interactions control regulatory T cell motility and immunological synapse formation.
J Immunol.
2014; 193(12): 5894–903. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 111.
Finlay DK, Rosenzweig E, Sinclair LV, et al.:
PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells.
J Exp Med.
2012; 209(13): 2441–53. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 112.
Dang EV, Barbi J, Yang HY, et al.:
Control of TH17/Treg balance by hypoxia-inducible factor 1.
Cell.
2011; 146(5): 772–84. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 113.
Shi LZ, Wang R, Huang G, et al.:
HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells.
J Exp Med.
2011; 208(7): 1367–76. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 114.
Ikejiri A, Nagai S, Goda N, et al.:
Dynamic regulation of Th17 differentiation by oxygen concentrations.
Int Immunol.
2012; 24(3): 137–46. PubMed Abstract
| Publisher Full Text
- 115.
Clambey ET, McNamee EN, Westrich JA, et al.:
Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa.
Proc Natl Acad Sci U S A.
2012; 109(41): E2784–93. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 116.
Winter SF, Minna JD, Johnson BE, et al.:
Development of antibodies against p53 in lung cancer patients appears to be dependent on the type of p53 mutation.
Cancer Res.
1992; 52(15): 4168–74. PubMed Abstract
- 117.
Gubin MM, Zhang X, Schuster H, et al.:
Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens.
Nature.
2014; 515(7528): 577–81. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 118.
Le DT, Uram JN, Wang H, et al.:
PD-1 Blockade in Tumors with Mismatch-Repair Deficiency.
N Engl J Med.
2015; 372(26): 2509–20. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 119.
Lathrop SK, Bloom SM, Rao SM, et al.:
Peripheral education of the immune system by colonic commensal microbiota.
Nature.
2011; 478(7368): 250–4. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 120.
Joshi NS, Cui W, Chandele A, et al.:
Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor.
Immunity.
2007; 27(2): 281–95. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 121.
Agne M, Blank I, Emhardt AJ, et al.:
Modularized CRISPR/dCas9 effector toolkit for target-specific gene regulation.
ACS Synth Biol.
2014; 3(12): 986–9. PubMed Abstract
| Publisher Full Text
- 122.
Toettcher JE, Weiner OD, Lim WA:
Using optogenetics to interrogate the dynamic control of signal transmission by the Ras/Erk module.
Cell.
2013; 155(6): 1422–34. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 123.
Guntas G, Hallett RA, Zimmerman SP, et al.:
Engineering an improved light-induced dimer (iLID) for controlling the localization and activity of signaling proteins.
Proc Natl Acad Sci U S A.
2015; 112(1): 112–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 124.
Victora GD, Schwickert TA, Fooksman DR, et al.:
Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter.
Cell.
2010; 143(4): 592–605. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 125.
Müller AJ, Filipe-Santos O, Eberl G, et al.:
CD4+ T cells rely on a cytokine gradient to control intracellular pathogens beyond sites of antigen presentation.
Immunity.
2012; 37(1): 147–57. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 126.
Moreau HD, Bousso P:
Visualizing how T cells collect activation signals in vivo.
Curr Opin Immunol.
2014; 26: 56–62. PubMed Abstract
| Publisher Full Text
Comments on this article Comments (0)