Keywords
Myosin, mechanosensing, mechanotransduction, migration, 3D, stress fiber, actomyosin bundle,
Myosin, mechanosensing, mechanotransduction, migration, 3D, stress fiber, actomyosin bundle,
The extracellular matrix (ECM) is a critical regulator of cell and tissue function. Properties of the ECM, including stiffness, topography, and ligand type and density, have all been shown to regulate cell shape, migration, and fate1,2. For example, matrix stiffness influences the differentiation of mesenchymal and neural stem cells into different lineages3–5. Substrate topography and stiffness can both direct cell migration and growth6–8. To effectively probe the properties of the ECM, the cell exerts forces on the environment and gauges the response in a controlled feedback loop that is broadly termed “mechanosensing”.
The cell has specialized machinery for ECM mechanosensing, including motor proteins, cytoskeletal proteins, and force-sensitive proteins that change conformation or activity (or both) in response to applied forces at focal adhesions (FAs), which are protein complexes that directly bind to ECM proteins through integrins and other ECM adhesion receptors9–11. In one important mode of mechanosensing, the cell uses stress fibers (SFs), which are bundles of 10 to 30 actin filaments in width12 (although some thicker SFs may contain up to ten times as many filaments in width) cross-linked by proteins, including α-actinin. Some SFs also contain non-muscle myosin II (hereafter referred to as MII), which lends contractile properties to the SF and enables the cell to survey ECM physical properties, define cell shape, and facilitate migration. This review will focus on recent advances in SF-based mechanosensing in both two-dimensional (2D) and three-dimensional (3D) environments.
MII has two important roles in SFs: (1) cross-linking antiparallel actin filaments and (2) generating the power stroke to translocate these filaments to contract the SF. MII is a hexameric protein complex composed of two myosin heavy chains, two essential light chains, and two regulatory light chains (RLCs) (Figure 1a). The heavy chains contain a helical tail domain and a globular head domain, which can bind to actin filaments and ATP13. Myosin complexes can further organize into bipolar filaments, with the tails in an antiparallel orientation and the actin-bound heads in opposing directions (Figure 1b). Polarized actin filaments are composed of actin monomers, which are polymerized onto the barbed (plus) end of an existing filament. To contract the filament, myosin heads hydrolyze ATP to generate rotation of the myosin head toward the plus end of actin, leading to the subsequent translocation of antiparallel actin filaments14.

(a) Myosin is composed of two heavy chains, each consisting of a globular head and a tail, two essential light chains, and two regulatory light chains. The non-helical tail region varies in the three isoforms. (b) Myosin heads bind to actin filaments. ATP hydrolysis leads to a conformational change in the head and neck region, which results in mechanical movement of the myosin head toward the plus end of actin and in movement of the actin filament in the opposite direction (indicated by arrows). (c) Stress fibers can be divided into three populations as defined by their anteroposterior position within a migrating cell and connection to focal adhesions. SF, stress fiber.
Actomyosin contractility is strongly regulated by phosphorylation of the RLC at Ser19 and Thr18. Ca2+-activated myosin light chain kinase (MLCK) and zipper-interacting protein kinase both phosphorylate the RLCs15–18. Additionally, Rho-GTPase effectors, including RhoA-activated Rho-associated kinase (ROCK) and p21-associated kinase (PAK), phosphorylate the RLCs14,18,19. ROCK can also reduce RLC dephosphorylation via inhibition of myosin light chain phosphatase activity14. Phosphorylation of Ser19 leads to an increase in Mg2+-ATPase activity that powers the MII head sliding along actin filaments and FA maturation15,17,20. Additional phosphorylation at Thr18 increases this activity and results in the clustering of actomyosin filaments into thick SFs16,20,21. The differential mechanical consequences of mono- versus di-phosphorylation remain an area of active study.
Three myosin isoforms—MIIA, MIIB, and MIIC—have been identified in mammalian cells, differing in their heavy chains. Expression of the three isoforms is not universal in cells. MIIA and MIIB are the predominant isoforms expressed in cultured cells, whereas MIIC is found in a more restricted subset of cells, including neural cells and breast and lung cancer cells14,22. In recently spread cells that have not yet established polarity, MIIA and MIIB uniformly co-assemble on the same SF23,24. Over time, as the cell becomes increasingly polarized, the leading edge becomes enriched in MIIA and the trailing end in MIIB25–27. Although SFs throughout the cell typically contain both MII isoforms, the ratio of MIIA to MIIB is higher in SFs near the leading edge, but decreases as SFs undergo retrograde flow during cytoskeletal remodeling23,28,29. This is likely due to a sorting mechanism driven by the different kinetics and heavy chains of the isoforms23. MIIA has a higher turnover rate and spends less time bound to actin compared with MIIB30,31. As SFs move in a retrograde manner, a higher proportion of MIIA unbinds from the fiber, which in turn enriches the SF in MIIB. Myosin chimeras consisting of swapped C-terminal tails reversed the localization of the isoforms32. These findings are consistent with the presumed differential functions of MIIA and MIIB. Rac1 promotes leading edge formation by generating a flat lamella and recruiting MIIA to the leading edge, where it quickly hydrolyzes ATP to form new, short-lived SFs30,33. MIIA also stabilizes adhesions and facilitates traction force generation at the leading edge34. On the other hand, MIIB has a slower ATP hydrolysis rate but a higher duty ratio, meaning that it spends more time bound to actin in its force-generating state, thereby generating higher force per ATP hydrolyzed31. This is important in stabilizing SFs, generating traction forces at the trailing edge, and maintaining the front-back polarity needed for directed migration23–26,35,36. Furthermore, MIIB is enriched in perinuclear SFs, where it compresses the cell nucleus to enable efficient cell migration and invasion through confined spaces37,38. MIIC is less well characterized; it is present in tumor cells and neural cells where it contributes to cytokinesis and neurite growth, respectively22,39,40.
To determine the minimal requirements for forming SFs, some have employed well-defined reconstituted systems consisting of purified filamentous actin and myosin to study the organization of actin and myosin into contractile bundles. Protein-level cues, including myosin concentration and actin polarity, guide the self-assembly and organization of myosin and actin filaments into contractile bundles, which are the building blocks of the tensed, interconnected SF network41–44. Analogous to the actomyosin bundles of differing actin polarities that form in reconstituted systems, SFs that vary in actin polarity have been observed in mammalian cells. Three populations of SFs—uniform polarity, graded polarity, and alternating polarity bundles, correlating with the intracellular location of the bundles—were first documented in migrating primary chick fibroblasts12. Uniform polarity bundles were observed near the cell front, and alternating polarity bundles were observed at the cell rear. Graded polarity bundles, in which the degree of polarity depended on the distance from the bundle ends, were located in the center of the cell12.
Recently, careful observation of SF dynamics in migrating U2OS osteosarcoma cells has given rise to a more general classification system for SFs on the basis of their different formation pathways, molecular composition, and connection to FAs (Figure 1c)45,46. Dorsal SFs are found at the lamella and have uniform actin polarity, due to inverted formin 2 or vasodilator-stimulated phosphoprotein (VASP) (or both) promoting actin polymerization at the barbed end (closest to the FA) of dorsal SFs45–48. Furthermore, they are often found to lack MII, implying that dorsal SFs are not contractile28,46,49. This subpopulation is connected at one end to an FA, and the other end rises toward the dorsal membrane surface. Dorsal SFs are mechanically coupled to the second subpopulation, transverse arcs. Transverse arcs are curved SFs exhibiting alternating actin polarity, and are found near the dorsal membrane surface of the lamella45. They are formed by the end-to-end annealing of Arp2/3-nucleated actin filaments and are not connected directly to FAs46,50. Transverse arc contraction, largely driven by MIIA activity, exerts a force on dorsal SFs in the retrograde direction. As dorsal SFs are anchored to the ECM via a stable FA, transverse arc contraction pulls dorsal SFs and the lamella membrane down28. The third subpopulation, ventral SFs, run along the matrix-bound face of the cell, become increasingly prominent toward the cell rear, and are connected at both ends to FAs. A subset of ventral SFs is produced from the myosin-mediated fusion of a transverse arc with two dorsal SFs46,48. Yet another classification system for SFs distinguishes between peripherally located SFs and centrally located SFs29,51–54. This scheme is motivated in part by the recognition that peripheral SFs (sometimes called peripheral arcs) can drive or reflect cortical surface tension (or do both) and that peripheral and central SFs can bear different mechanical loads54–56.
The primary chick fibroblast SF classification system can perhaps be reconciled with the U2OS SF classification system. The uniform polarity bundles and alternating polarity bundles correspond to dorsal and ventral SFs, respectively. The graded polarity bundles correspond to the transverse arcs fusing with dorsal SFs on either side during retrograde flow46. The degree of polarity corresponds to the location of the SF within a migrating cell. At the lamella, SFs undergoing active and directed polymerization have uniform polarity in order to stabilize the protrusion of the leading edge. As the SFs move toward the trailing edge of the cell, SFs adopt an alternating polarity, indicating that their primary role is to generate contractile forces to maintain cell shape and traction. Peripheral SFs can be classified as ventral SFs (or multiple ventral SFs bundled together), and central SFs can broadly encompass dorsal SFs, transverse arcs, and ventral SFs.
It is important to note that the dorsal/transverse arc/ventral SF was originally developed for mesenchymally migrating cells and that the uniform/graded/alternating polarity system was based on observations in primary chick fibroblasts. The peripheral/central SF classification scheme is the most general and is applicable to many cells. Not all cell types exhibit the dorsal/transverse arc/ventral SF subpopulations, and even within the same population of cells, there may be variability in the representation of each of the SF subpopulations28,45,46. Stationary cells often exhibit only ventral SFs, indicating that one of the primary roles of dorsal SFs and transverse arcs is to drive leading edge protrusion during migration. The varying degrees of SF representation raises the question of how different ECM cues, including stiffness, ligand presentation, and dimensionality, collectively influence SF subpopulation formation and organization. Furthermore, there are other questions pertaining to the how SF subpopulations interact to form an interconnected network. For example, transverse arc-dorsal SF junctions are not well characterized at the molecular scale but are likely enriched in actin cross-linking proteins that promote force transmission by tightly coupling dorsal SFs to transverse arcs. These areas are currently under active investigation.
It is widely appreciated that MII tenses SFs to different degrees in cells. Measurements of the tensile properties of actomyosin bundles have been carried out on reconstituted actomyosin systems or isolated SF networks where all other cell components are removed57. In these simplified systems, SFs can be manipulated to measure their biophysical properties by using tools, including microcantilevers58. However, these methods are not amenable to live cells. Thus, to study SFs in live cells, some have used outside-in perturbations to measure mechanical properties of SFs, including nanoindentation and whole-cell stretching59,60. Others have used inside-out methods such as pharmacological treatment or genetic perturbations to manipulate SF architecture and tension and measure the resulting changes in the ability of the cell to exert traction on the ECM50,51,61,62. However, with these methods, it is not possible to tease out the mechanical contributions of individual SFs and to examine how they contribute to the overall contractility of the cell.
Thus, our group29,52–54 and others63 have used femtosecond laser nanosurgery to sever single SFs to directly measure the mechanical properties, including contractility, of SFs within living cells and confirmed the presumed cross-linking and contractility roles of MII. When ventral SFs are severed, the cut ends retract in a viscoelastic manner which is largely mediated by MII52,53. MII cross-linking imparts viscous resistance to retraction of a severed SF, as deletion of the actin-binding myosin head speeds SF retraction29. At the same time, MII activity contributes to SF elasticity by tensing actin filaments. The retraction kinetics of SFs differ based on the location of the SF: peripheral SFs retract a longer distance and with a lower effective elasticity (longer time constant) than centrally located SFs, indicating that peripheral SFs are tensed to a greater degree53. These differences may be associated with the spatially compartmentalized control of myosin RLC kinases. Peripheral SFs are preferentially regulated by MLCK, and central SFs by ROCK, as pharmacological inhibition of the kinases using ML-7 (MLCK) or Y-27623 (ROCK) affected the retraction kinetics and morphology of the respective populations51,64. Some studies suggest that the ratio of MIIA to MIIB isoforms on a particular SF can affect its mechanical properties and that ROCK preferentially regulates MIIA activity whereas MLCK preferentially regulates MIIB29,32,65. These findings may be placed in the context of the different mechanochemical properties of MIIA and MIIB. In particular, ROCK-controlled SFs may be enriched in fast ATP-hydrolyzing MIIA, which facilitates the rapid and dynamic SF contraction and evolution in the lamella. MLCK-controlled peripheral SFs may be enriched in high-duty ratio MIIB to support the stable SFs found at stable cell edges. However, additional studies are needed to test these hypothetical associations in a clear and direct way and to examine the differential mechanics of the various SF subpopulations. It would be particularly interesting and important to relate the changes in SF composition and regulation in specific cellular compartments to mechanical functions.
MII plays a critical role in sensing mechanical properties of the ECM, including stiffness, by exerting traction stresses on the substrate1,9,66,67. On softer substrates, FAs are smaller and SFs are less abundant as cells are unable to generate sufficient traction that would otherwise reinforce adhesions66,68,69. The diminished traction forces can restrict cell spreading and migration, and in some cases are associated with reduced proliferation7,8,68,69. In contrast, cells are able to generate large traction stresses on stiff substrates, which enable them to spread and form mature FAs66,67. The differences in morphology between cells cultured on compliant and non-compliant substrates are understood to be MII-mediated, because cells lose their characteristic stiffness-dependent differences with abrogation of myosin-based contractility8,70. Although this review focuses on MII, there are also several other classes of myosin motors whose roles in mechanosensing are under investigation. These myosin motors typically bridge actin filaments to other proteins. For example, myosin X, which links actin to membrane proteins, is critical in the formation of filopodia, thin actin protrusions that participate in ECM remodeling71–73. In turn, filopodia may contribute to the formation of dorsal SFs74. Future experiments should uncover the roles of other myosin motors in mechanosensing.
Within a given cell, different pools of SFs appear to exert different levels of traction. Although this idea is still being systematically explored, computational analysis of experimental data offers important clues. For example, model-based traction force microscopy (TFM) infers tension held in SFs by iteratively matching traction maps and images of SFs and FAs with cable network models of the actin cytoskeleton75. These measurements reveal that individual ventral SFs exert the highest traction forces and dorsal SFs exert the lowest75. More conventional TFM studies suggest that dorsal SFs are more important for templating the location of adhesions and rely upon MII activity in the cortical actin cytoskeleton (for example, transverse arcs) to drive force-dependent FA growth61. Interestingly, although the traction force per dorsal SF is relatively low, the lamellipodium, which lacks defined SFs, can generate very high traction forces that seem to be largely driven by cortical MII activity76,77. SF-generated traction likely becomes more important in generating traction forces and defining cell shape in areas further away from the lamellipodium. Dorsal SFs, which are found behind the lamellipodium, directly interact with FAs but can neither generate contractile forces nor exert traction on their own since they lack MII. Instead, they exert low traction forces indirectly through transverse arc contractility. Ventral SFs are the predominant SF type in non-migrating cells, which by definition lack front-back polarity. They are under higher tension and generate higher traction forces than either dorsal SFs or transverse arcs75. Peripherally located ventral SFs collectively exert higher traction stresses compared with centrally located ventral SFs52,54.
Individual tensed SFs are networked together to form a dynamic system that can readily redistribute tension54. Femtosecond laser nanosurgery is a powerful tool that can be used to obtain mechanical properties of selected SFs and their role in maintaining tension redistribution. For example, a single SF can be severed to elucidate its structural role in the cytoskeleton by examining changes in SF morphology in the surrounding network. Combining this technique with molecular readouts, such as Förster resonance energy transfer (FRET) tension sensors (for example, based on vinculin78, talin79, or α-actinin80), may provide insight into how tension released from a single SF is balanced by the surrounding cytoskeletal network.
Mechanosensing by MII pulling on FA-anchored SFs has been described by the motor-clutch model. In this model, MII and FAs respectively act as a cellular motor and clutch mechanism that can probe substrate stiffness and direct actin polymerization9. Spreading cells initiate stiffness sensing by locally tensing the substrate through sarcomeric units consisting of a myosin minifilament (comprised of about 28 myosins arranged in a bipolar fashion) cross-linked to two actin filaments which in turn are connected to nascent focal complexes81. The ECM stiffness value correlates with the number of steps the MII motors take (roughly 2.5 nm per step) before the actin filaments reach a force threshold required to recruit proteins to reinforce and stabilize the adhesion81. Stiff substrates require fewer myosin miniflament steps to recruit and promote nascent adhesions into stable FAs81. These adhesions then associate with SFs and are integrated into the cytoskeletal network, which results in increased tension on the adhesion. These forces unfold mechanically sensitive FA proteins (including talin and vinculin) and, in a positive feedback loop, initiate signaling cascades that produce thicker and highly tensile SFs10,82–84. During this process, frictional slippage occurs, whereby actin moves relative to the stationary FA9. Conversely, on more compliant substrates, the myosin miniflaments within a sarcomeric unit are required to take a larger number of steps to reach a force threshold. Load-and-fail dynamics, where the ECM-coupled nascent adhesion moves with the actin filament until a failure point is reached and the adhesion detaches from the ECM, may also be observed9,11,85–88. In this regime, the rate of integrin disengagement from fibronectin, an ECM ligand, is faster than the rate of talin unfolding, which precludes vinculin binding and FA reinforcement10. This results in cells with thinner SFs (or no SFs at all) and cells with smaller projected areas68,89,90.
The cytoskeleton undergoes continuous remodeling in response to changes in the environment. When an SF is under tension, VASP is phosphorylated along the SF, leading to increased contractility and a cessation of actin polymerization at the FAs48. It is conceivable that a sarcomeric force-sensing mechanism similar to the one described above at the cell-ECM interface also exists along the length of SFs. That is, on stiff substrates, MII minifilaments would need to take a small number of steps along filaments to reach a threshold force. Increasingly stiff substrates favor the addition of sarcomeric units along the length of the fiber, which incrementally lengthens the SF. This suggests that longer SFs with more sarcomeric units bear more tension. On the other hand, when an SF is no longer under a threshold tension, VASP is not phosphorylated and the SF is targeted for disassembly by cofilin48. These two mechanosensitive mechanisms provide a mechanism for stiffness sensing and durotaxis at the FA and SF: nascent adhesions or SFs that are not under a threshold tension are disassembled, leaving behind stable SFs and adhesions.
MII activity and the ability to sense stiffness cues in the environment mediate various aspects of tumor progression, including dysplasia, tissue invasion, and metastasis19. When manipulated in culture, matrix stiffness and ligand density both affect the ability of cells to migrate, and migration speed is maximized at intermediate levels of both91. These biphasic relationships have been successfully explained by using models that involve myosin-based mechanosensing92,93. Furthermore, the orientation of matrix proteins, including collagen and fibronectin, determines the ability of cells to effectively engage with the ECM during mechanosensing94–96. Aberrant mechanosensing has been implicated in the pathogenesis of diseases involving cell migration through tissue, including the invasive brain tumor glioblastoma (GBM). Whereas soft matrices reduce the migration of GBM cell lines in a MII-dependent fashion8, primary GBM tumor-initiating cells spread, migrate, and proliferate even on very compliant matrices97. Increasing myosin contractility through pharmacological or genetic manipulation restores the expected loss of motility, spreading, and proliferation on compliant substrates and dramatically reduces invasion in vivo97. Interestingly, myosin activation has also been observed to facilitate GBM cell translocation through tight intercellular spaces within the brain37. Future studies should uncover how the reported in vitro roles of MII can be translated into disease microenvironments.
Most studies of myosin-mediated SF regulation of cell shape have been conducted in cells cultured on idealized 2D substrates with the basal side interacting with the ECM-coated surface and the dorsal side free. Many of these studies highlighted above focused on one aspect of the microenvironment, such as adhesivity or stiffness, whereas the in vivo microenvironment, which is often 3D, can vary in pore/mesh size, degradability, geometry, stiffness, and protein composition. Recent efforts have focused on better understanding the role of complex matrices that are more representative of the in vivo conditions, such as interfacial 2D and fully 3D environments. For example, during invasive migration along a blood vessel-ECM interface, tumor cells interact with blood vessels on the basal surface and ECM proteins on their dorsal surface98,99. In fully 3D environments, cells are often embedded in a meshwork of ECM proteins (for example, collagen) and may interact with several fibers in different planes. The additional dimension introduces another degree of freedom that can significantly alter migration and cell shape from a slowly migrating, lamellipodial shape to a fast migrating, elongated shape100. The role and existence of SFs in vivo have been controversial, as they are sometimes assumed to be an artifact of 2D culture101,102. However, recent publications indicate that contractile SFs are important in vivo in processes as varied as wound closure, embryonic epithelial sheet closure, and duct contraction103. Cells also form SFs in 3D matrices consisting of thick collagen bundles62,104. It is unclear whether 3D SFs, which are often thinner and more difficult to visualize using conventional confocal microscopy techniques, can also be described by the dorsal/transverse arc/ventral SF classification scheme for 2D cultures. Super-resolution imaging and femtosecond laser ablation may be used to better understand the structure, composition, and mechanical properties of these 3D SFs.
To better understand the roles of actomyosin contractility and migration in complex systems and to compare the roles in 2D environments, researchers have used different culture systems to replicate in vivo conditions. To mimic interfacial migration, we98 and others105 have developed 2.5D sandwich systems that confine cells between a planar base substrate and an ECM or hydrogel layer. In these systems, cell migration is slower and morphology becomes elongated with no lamellipodia, in the case of GBM cells98. This is attributed to the ECM overlay, which promotes the formation of additional adhesions on the dorsal surface. MII inhibition prevents the formation of strong adhesions to both surfaces and thus enables the cell to migrate faster98.
Others have also embedded cells in collagen matrices to mimic 3D ECM environments. At the macroscale, collagen forms a soft gel with a 1 kPa Young’s modulus, which is very different from the microscale structure consisting of long fibers with megapascal-scale Young’s modulus (measured from the long axis) that single cells effectively sense104. As in the sandwich cultures, cells adopt an elongated spindle morphology in these matrices and align their adhesions and SFs along collagen fibers94,98,104. The local fiber architecture is critical in determining adhesion size and ultimately the magnitude of traction forces that the cell can exert (Figure 2)94. Collagen has a megapascal-scale tensile strength along the long axis but a much smaller stiffness if measured in the normal direction. Thus, FA area is larger if cell-generated forces are applied parallel to the fiber and smaller if applied normally94,106.
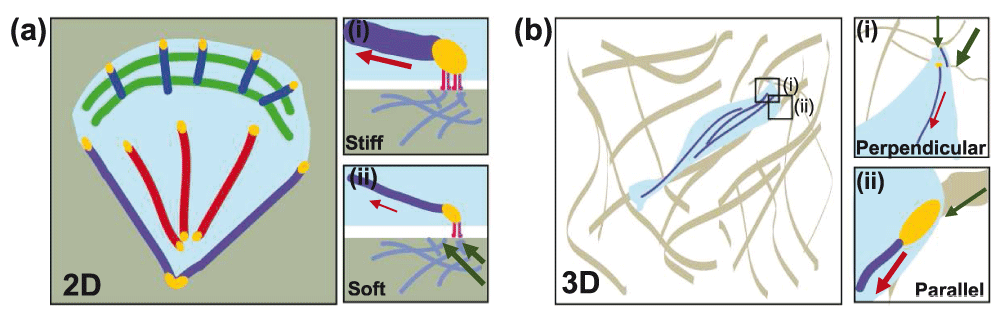
(a) Migrating cells on two-dimensional matrices have a broad, flat leading edge and a pointed trailing end. Dorsal SFs are in blue, transverse arcs in green, ventral SFs in red, and peripheral SFs in purple. Focal adhesions are in yellow. (i) Stiff substrates are able to resist deformation by cell-generated forces (red arrow). This results in focal adhesion maturation and reinforcement of SFs. (ii) Soft substrates deform (green arrows) under cell-generated forces and move with the applied force. Focal adhesions are smaller and SFs are thinner. (b) In three-dimensional collagen matrices, cells adopt an elongated morphology. Collagen fibers have high tensile strength but low resistance to bending. (i) Fibers oriented normally to the cell-generated force (red arrow) readily deform (green arrows). (ii) Fibers oriented parallel to the applied force (red arrow) are tensed (green arrow) and support the formation of mature adhesions and SFs.
Given that collagen microarchitecture varies in stiffness and pore size, we93,108 and others38,100,109 have engineered well-defined matrices to decouple these two parameters. We developed a polyacrylamide microchannel platform in which substrate stiffness and confinement can be independently varied. In these environments, cells in stiff, thin-width microchannels form SFs along the channel walls and migrate faster compared with those in soft, wider channels93,107,108. The effects of stiff, thin-width channels on cell morphology and migration speed are consistent with those observed in collagen matrices. Others have also fabricated microfluidic devices featuring constrictions of varying widths and found that MIIB is responsible for squeezing the rigid nucleus through these environments38,109. In yet another approach, thin-width, high-aspect ratio patterned fibronectin strips were used to examine the effects of topography and ligand density on migration and morphology. These “1D” photopatterned strips are reminiscent of the thin fibrillar collagen tracks that cells migrate along in the 3D collagen matrices. In contrast to cells on 2D substrates, cells cultured on the 1D systems have elongated spindle morphologies and fast migration speeds similar to those observed in fibrillar 3D matrices100. Furthermore, unlike in 2D, where the correlation between migration speed and ligand density is biphasic, migration speed is independent of ligand density in 1D100.
Efforts to understand the differential roles of MII isoforms in non-2D systems have also yielded surprising results. MIIA is required to stabilize adhesions and form a flat lamella in 2D and is also required for FA maturation at the leading edge of cells in 1D photopatterned ECMs28,34,110. MIIB is required to stabilize mature adhesions further back from the leading edge25,27,110. However, inhibition of MII activity has different effects on cells in 2D and 3D. In 2D, genetic ablation of MIIA or pharmacological inhibition of MII activity increases mesenchymal migration speeds, whereas in 3D, migration is abrogated34,104,110,111. This effect is likely due to differences in migration modes: 2D mesenchymal migration is a slow process that is dependent on the formation and maturation of adhesions. Inhibition of MII increases migration speeds by increasing FA turnover and preventing their maturation, which impedes efficient migration98. There is also a possibility that MII inhibition increases the actin monomer pool (which would otherwise be incorporated into thick SFs) at the leading edge, allowing actin polymerization to promote leading edge protrusion for migration. In contrast, in 3D, MII inhibition effectively abrogates migration since actomyosin contractility is needed to break the high levels of integrin clustering that are found in 3D matrices104. Migration by actin polymerization-driven leading edge protrusion is limited since the discontinuous fibers are much smaller in area compared with the 2D case.
In vivo, the ECM is highly complex and variable in stiffness, dimensionality, and ligand presentation. These different combinations of matrix properties may influence cell behavior in complex and unpredictable ways that are challenging to deduce from studies in which single properties are varied in isolation. Although it seems clear that MII-mediated actomyosin contractility within SFs plays crucial roles in mechanosensing in 2D culture, the field is still grappling with the translation of these relationships to more complex microenvironments representative of tissue. Thus, an important objective going forward will be to characterize these relationships, which will surely be facilitated by developing more sophisticated culture paradigms.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)