Keywords
Leukocytes, endothelial barrier, transmigration, diapedesis
Leukocytes, endothelial barrier, transmigration, diapedesis
Leukocytes circulating in the bloodstream represent a reservoir of immune cells that are passive as long as they are circulating. To bring them into a position where they can perform their immune functions, they need to first exit the vascular system. This requires sophisticated mechanisms that allow the leukocytes to recognize injured or infected tissue areas from within the vasculature. Recognition goes hand in hand with adhesion to the luminal surface of endothelial cells. These initial events are mediated by cytokine-induced endothelial adhesion molecules, such as the selectins, that mediate the capturing and rolling of leukocytes at the vessel wall. Selectins and chemokines presented on the endothelial cell surface trigger the activation of leukocyte integrins, which initiates leukocyte arrest and supports the crawling to appropriate exit sites. This well-studied multi-step process of leukocyte docking has been described in several excellent reviews1–5.
Here, we will focus on recent advances in our understanding of the subsequent steps of leukocyte extravasation, which are less well understood. For more extended discussions, the reader is also referred to some other recently published excellent reviews6–9. We will first give a short overview about endothelial adhesion receptors and signaling processes that support the diapedesis process. Considering this, we will then discuss what is currently known about the following questions: what determines the exit sites where the transmigration (diapedesis) process occurs and how do leukocytes recognize such sites? Which routes do leukocytes take to transmigrate through the endothelial barrier—the paracellular pathway through junctions or the transcellular route through the body of an endothelial cell—and what determines which route is taken? How are paracellular or transcellular gaps or pores through the endothelial barrier formed? How do leukocytes and endothelial cells maintain the barrier integrity and prevent plasma leakage during the diapedesis process?
After capturing, rolling, and arrest, leukocytes crawl on the endothelial surface until they start to diapedese through the endothelial barrier. Arrest and crawling are mediated mainly by the β2-integrins LFA-1 (αLβ2) and Mac-1 (αMβ2) and the β1-integrin VLA-4 (α4β1) on leukocytes, of which the first two bind to endothelial intercellular adhesion molecule 1 (ICAM-1) and the latter binds to vascular cell adhesion protein 1 (VCAM-1). These endothelial adhesion molecules also act as signaling receptors and are instrumental for the initiation of signaling events, which affect the actomyosin cytoskeleton as well as adhesive structures at endothelial junctions, thereby facilitating and enabling the transmigration process (Figure 1A). The concept that leukocytes trigger endothelial cells to support leukocyte transmigration goes back to studies by the Silverstein lab, which showed that neutrophil binding to endothelial cells triggered a Ca2+ signal inside endothelial cells, which was required for transmigration but not for leukocyte adhesion10. Later, this was linked to the phosphorylation of myosin light chain and the induction of isometric tension in endothelial cells11. The relevance of Ca2+ transients for the diapedesis process was confirmed in several reports12,13, although differences were found depending on which type of leukocytes were analyzed and whether endothelial cells were activated with cytokines14. ICAM-1 was identified as a receptor that triggered lymphocyte-induced Ca2+ transients in endothelial cells13, and E- and P-selectin and VCAM-1 were also reported to have this capacity15. More recently, it was found that the Ca2+ channel TRPC6 is responsible in endothelial cells for Ca2+ transients that are induced by leukocytes and are required for transmigration (Figure 1A)16. Another ion channel in endothelial cells, which is important for the recruitment of T cells into the brain, is the TWIK-related potassium channel-1 (TREK-1)17.
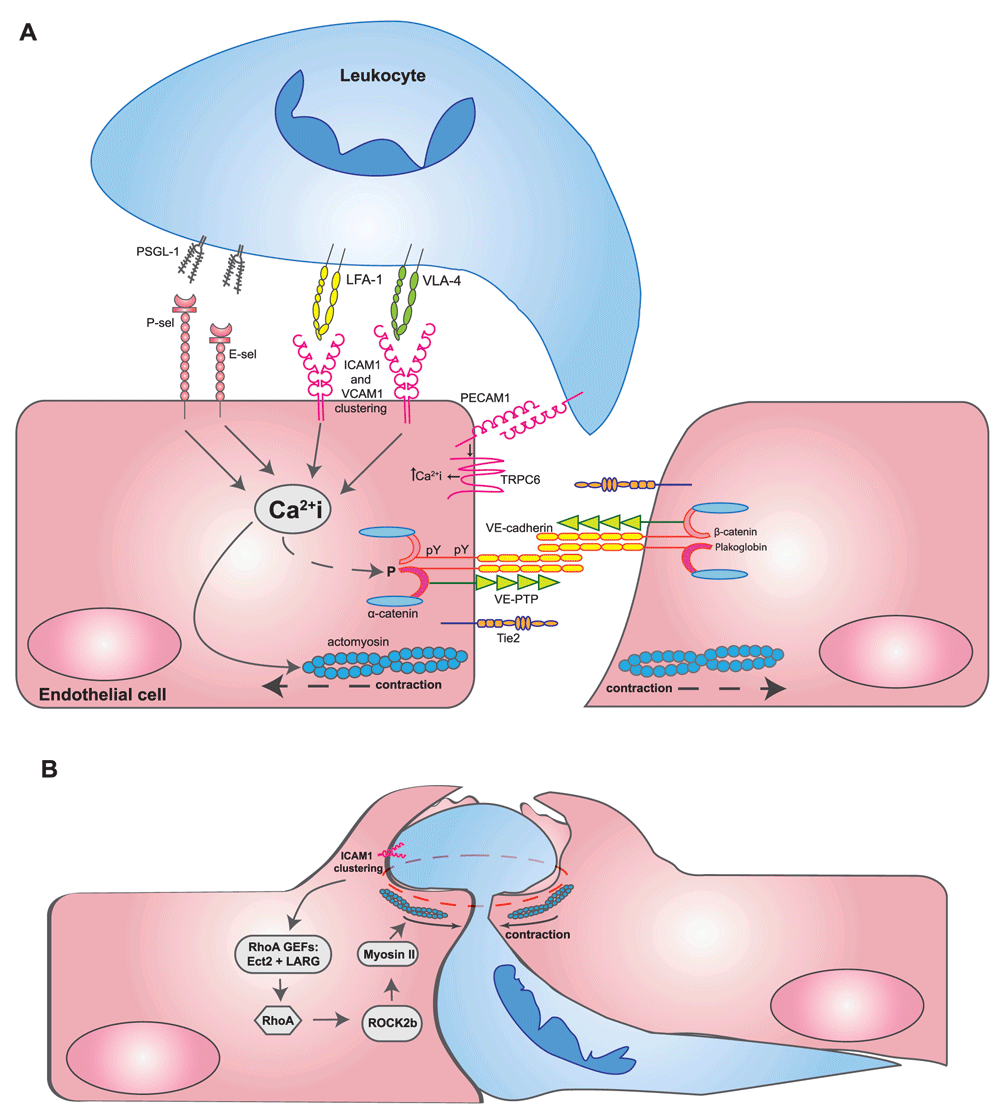
(A) Leukocytes interacting with several adhesion molecules on the endothelial cell surface trigger Ca2+ signals inside endothelial cells, which are essential for leukocyte transmigration. It was reported that Ca2+ signals triggered by the apical adhesion molecules were initiated by stores from the endoplasmic reticulum but that PECAM-1 Ca2+ transients occurred rather local at transmigration sites through the TRPC6 channel. Ca2+ signals trigger the activation of actomyosin-mediated pulling on endothelial junctions, influence the phosphorylation of components of the VE-cadherin-catenin complex, and trigger the recycling of the lateral border recycling compartment (LBRC) vesicle compartment. For a more detailed depiction of intracellular signaling steps, the reader is referred to recent reviews7–9. (B) When leukocytes have already transmigrated more than halfway through the site of diapedesis, RhoA-mediated signaling triggered by the RHO guanine nucleotide exchange factors (GEFs) Ect2 and LARG stimulates ROCK2b, which activates actomyosin-based forces that support pore confinement, which leads to closure of the diapedesis pore87. Abbreviations: ICAM1, intercellular adhesion molecule 1; LFA-1, lymphocyte function-associated antigen 1; PECAM-1, platelet and endothelial cell adhesion molecule 1; PSGL-1, P-selectin glycoprotein ligand-1; TRPC6, transient receptor potential canonical-6; VCAM1, vascular cell adhesion molecule 1; VE-cadherin, vascular endothelial cadherin; VE-PTP, vascular endothelial protein tyrosine phosphatase; VLA-4, very late antigen-4.
Other signaling steps that are triggered by ICAM-1 and VCAM-1 are summarized in excellent reviews18,19. Activation of Rho family kinases such as RhoG is involved in the stable adhesion of leukocytes to the endothelium20. In addition, the activation of RhoA is involved in ICAM-1-dependent rearrangements of the actomyosin system, leading to mechanotransduction in endothelial cells, which supports leukocyte transmigration21–24. Stimulation of the activation of Src family kinases leads to tyrosine phosphorylation of cortactin, which is relevant for the clustering of ICAM-125–27. Furthermore, clustering of ICAM-1 modulates tyrosine phosphorylation of vascular endothelial cadherin (VE-cadherin) and associated catenins, which is important for the opening of endothelial junctions28–30.
Several endothelial membrane proteins support the transmigration of leukocytes through the endothelial cell barrier. Platelet and endothelial cell adhesion molecule 1 (PECAM-1) was the first adhesion molecule that was shown to be involved in this process31 and was followed by many others such as the junctional adhesion molecules JAM-A, -B, and -C32–35; endothelial cell-selective adhesion molecule (ESAM)36; CD9937,38; CD99L239–41; and the nectin-related poliovirus receptor (PVR)42. All of these proteins are enriched at endothelial cell contacts. Although not much is known about how they facilitate the diapedesis process, some of them were shown to act in a sequential manner. PECAM-1 was found to act before CD9937, whereas PVR functions after PECAM-1 and before the CD99 step42. ICAM-2 was reported to support one of the earliest steps in the diapedesis process followed by JAM-A and then PECAM-143. Interference with some of these receptors, such as PECAM-1, CD99, and CD99L2, led to the accumulation of leukocytes between the endothelium and the basement membrane, suggesting that these proteins were also involved in mechanisms that enable leukocytes to overcome the basement membrane44,45.
Mechanistically, it was suggested that a multi-vesicular compartment inside endothelial cells, the lateral border recycling compartment (LBRC), would support the diapedesis process, possibly by serving as a membrane reservoir that helps accommodate the body of the transmigrating leukocyte at gaps between endothelial cells46. PECAM-1 was suggested to trigger the mobilization of this compartment to the plasma membrane, whereas crosslinking of CD99 would trigger a second wave of vesicle traffic to cell contacts47. The latter process was linked to the interaction of CD99 with ezrin, soluble adenylyl cyclase, and protein kinase A (PKA)47. Engagement of PECAM-1 by leukocytes was reported to cluster the Ca2+ channel TRPC6 at sites of diapedesis, and TRPC6 was found to act downstream of PECAM-1, affecting the recycling of LBRC vesicles16. This report also suggested that Ca2+ signals triggered by PECAM-1 via TRPC6 were rather local and different from global cellular Ca2+ signals that are found after leukocyte docking to endothelial cells, which are mediated by Ca2+ store release from the endoplasmic reticulum (Figure 1A).
Crawling on the luminal surface of postcapillary venules was reported to help leukocytes find appropriate exit sites48. Several mechanisms were recently discussed which could determine such exit sites and allow leukocytes to identify them. Since ICAM-2 seems to act at the most apical position of all diapedesis-supporting cell surface receptors43, it might be a candidate for a receptor that could help leukocytes to identify endothelial cell contacts. ICAM-2 is diffusely expressed over the whole apical surface of endothelial cells in postcapillary venules but is enriched at endothelial cell contacts49. Apical ICAM-2 supports as a ligand for Mac-1 neutrophil crawling and interfering with its function altered the stop-and-go intervals of leukocyte crawling49. It is an attractive speculation that the increased expression levels of ICAM-2 at cell contacts might influence leukocytes in their decision to stop and start diapedesing.
Recently, platelets were described as pathfinders for leukocyte extravasation. It was shown that platelets adhered under inflammatory conditions at endothelial junctions in the smallest venular microvessels and captured neutrophils via CD40-CD40L/CD154-dependent interactions50. Intravascularly adherent platelets and neutrophils, in turn, recruited inflammatory monocytes to these sites of extravasation. These interactions required the interaction of P-selectin with leukocyte PSGL-1, which contributed to the activation of leukocyte integrins. Blockade of these multi-cellular interactions reduced leukocyte extravasation. These findings provide mechanistic understanding for the previously well-documented important contribution of platelets to leukocyte extravasation51–54.
Another aspect that may determine a transmigration site is the stiffness of endothelial cells. It was found that endothelial stiffness supported the spreading and transmigration of neutrophils. A gradient of increasing stiffness (measured by atomic force microscopy) from the center to the periphery of endothelial cells drove crawling neutrophils toward cell junctions, promoting transmigration through a paracellular route55. In contrast, another study showed that lymphocytes transmigrated preferentially through local sites of reduced endothelial cell stiffness, which were characterized by low levels of F-actin and were also found at sites of transcellular migration56. Interestingly, long time exposure of endothelial cells to flow stabilizes junctions and this increased the fraction of lymphocytes that transmigrated through a transcellular route56.
Leukocytes transmigrate through a paracellular route, which requires the opening of endothelial junctions, and through a transcellular route, which does not require junction opening but is often close to junctions. Both processes have been well documented in vitro and in vivo. Direct analysis by intravital three-dimensional video microscopy showed that 90% of extravasating neutrophils use the paracellular route and that only 10% use the transcellular route, and this was found under different inflammatory stimuli35. In line with this, stabilizing endothelial junctions in genetically modified mice by replacing endogenous VE-cadherin with a VE-cadherin-α-catenin fusion construct strongly inhibits neutrophil extravasation in lung and cremaster and lymphocyte recruitment into inflamed skin by 70 to 80%57. In vitro, more than 90% of neutrophils, monocytes, and lymphocytes transmigrate through human umbilical vein endothelial cells (HUVECs) via the paracellular route58. The low efficiency of the transcellular route is enhanced up to 30% for leukocyte transmigration through cultured microvascular endothelial cells59. In addition, higher expression levels of ICAM-1 on endothelial cells correlated with a higher percentage of transcellular events60,61. The latter study also showed that higher ICAM-1 expression levels correlated with reduced crawling distances of cells under flow. Interestingly, this study also reported that the few lymphocytes that still transmigrated through endothelial cells lacking ICAM-1 and ICAM-2 were unable to crawl prior to diapedesis and used exclusively the transcellular route61. Thus, it is tempting to speculate that leukocytes, which are hindered to reach their optimal site of exit at junctions or which reside longer than normal at a site on the apical endothelial surface, may tend to use a transcellular route. In agreement with this concept, interfering with the integrin Mac-1 on neutrophils, which inhibited crawling, enhanced the percentage of transcellular migration in vivo48. Likewise, T cells deficient for the Rac1 guanine nucleotide exchange factor (GEF) Tiam1 showed reduced crawling and used the transcellular diapedesis route with enhanced efficiency62.
Paracellular diapedesis of leukocytes requires the opening of endothelial junctions. VE-cadherin is an important player in this process since antibodies against VE-cadherin can enhance leukocyte extravasation in vivo63 and enhancing the adhesive activity of VE-cadherin by replacing it with a VE-cadherin-α-catenin fusion protein in knock-in mice strongly inhibits leukocyte extravasation in several tissues57. In vitro studies revealed that leukocyte-triggered clustering of ICAM-1 modulated tyrosine phosphorylation of VE-cadherin, which was linked to the opening of junctions and transmigration efficiency28,29. The transmigration of B16 melanoma cells through cultured endothelial cells was reported to require endothelial FAK activity, which triggered the phosphorylation of Y658 of VE-cadherin64. In vivo studies with mice carrying tyrosine/phenylalanine (Y/F) point mutations in Y731 or Y685 of VE-cadherin revealed that leukocyte-induced dephosphorylation of Y731 was required for neutrophil extravasation in vivo30 (Figure 1A). This dephosphorylation was dependent on the phosphatase SHP-2, which led to enhanced endocytosis of VE-cadherin. Interestingly, stimulation of vascular permeability by inflammatory mediators did not require Y731 of VE-cadherin but was dependent on the upregulation of pY68530. This revealed that opening of endothelial junctions in vivo requires the modulation of VE-cadherin tyrosine phosphorylation; however, different tyrosines are addressed in the context of vascular permeability induction and leukocyte extravasation. It is attractive to speculate that the dephosphorylation of Y731 on VE-cadherin initiates a more robust opening of junctions, which allows the passage of transmigrating leukocytes, whereas the induction of Y685 phosphorylation leads to a more subtle destabilization of endothelial contacts, which is sufficient for plasma leaks.
An important regulator of endothelial junction integrity is the endothelial-specific receptor-type tyrosine phosphatase called vascular endothelial protein tyrosine phosphatase (VE-PTP), which associates with VE-cadherin and supports its adhesive activity65, in part by inhibiting tyrosine phosphorylation of plakoglobin66. Docking of leukocytes to endothelial cells as well as stimulation of endothelial cells with vascular endothelial growth factor (VEGF) or histamine triggers rapid dissociation of VE-PTP from VE-cadherin66. Each of these different stimuli triggers the same signaling cascade that leads to VE-PTP/VE-cadherin dissociation67. It could be demonstrated that this dissociation is necessary for the induction of vascular permeability and for inflammation-induced neutrophil extravasation in vivo68. Interestingly, VE-PTP is able to dephosphorylate Y685 but not Y731 of VE-cadherin30. Thus, it is likely that VE-PTP affects the regulation of vascular permeability via Y685 of VE-cadherin but that the substrate relevant for the role of VE-PTP in leukocyte diapedesis is probably plakoglobin30,66. A role of VE-PTP for the control of endothelial cell integrity in vivo was also shown in a recent study that reported the induction of VE-PTP by hypoxia via HIF2α69. Besides VE-cadherin, VE-PTP also associates with the tyrosine kinase receptor Tie-270, which is important for vascular remodeling in embryonic development71 (Figure 1A). Interfering with VE-PTP in various ways leads to the stabilization of endothelial junctions72. This effect is mediated by Tie-2. It also attenuates neutrophil recruitment into inflamed tissue by blocking actomyosin pulling forces on endothelial junctions73.
In addition to counteracting the function of VE-cadherin, leukocytes also need to trigger mechanisms that actively open gaps for extravasation. As mentioned above, clustering of ICAM-1 triggers endothelial Ca2+ signals and phosphorylation of myosin light chain kinase (MLCK)11, which has been implicated in the modulation of endothelial junctions74. Inhibition of MLCK prevents neutrophil diapedesis75, and also Rho kinase was shown to be involved76. In agreement with this, the endothelial microfilament system is required for the transmigration of monocytes77. These reports are in line with our previous findings that showed that stimulation of the endothelial tyrosine kinase receptor Tie-2 can inhibit neutrophil recruitment to endotoxin-stimulated mouse lungs73. These effects were even observed in mice conditionally gene-inactivated for VE-cadherin. Since Tie-2 activation was found to reduce radial stress fiber formation and blocked MLC phosphorylation by a Rap-1- and Rac-1-dependent mechanism, these results suggest that leukocyte extravasation opens endothelial junctions by a two-step mechanism: downregulation of VE-cadherin function and active, actomyosin-mediated pulling on endothelial cell contacts73.
For the transcellular migration pathway, it is less clear how the required transcellular gaps are formed. It was shown that membrane trafficking-related proteins such as vasodilator-stimulated phosphoprotein (VASP) and caveolin are involved in the formation of the transcellular pathway78. Furthermore, it was suggested that regulated membrane fusion events requiring NSF (N-ethylmaleimide sensitive factor) and SNARE (soluble NSF attachment protein receptor) complex proteins would be required in endothelial cells for transcellular leukocyte diapedesis59. Endothelial cells are often rather flat in large areas of their bodies, with a thickness of no more than 1 μm at their edges. It may be possible that at certain sites the apical and the basal membranes fuse directly. Alternatively, intracellular vesiculo-vacuolar organelles inside endothelial cells might fuse with each other and with the apical and basal membranes to form a short “channel” that could accommodate transmigrating leukocytes on a transcellular route79. It is interesting that several of the adhesion receptors that are involved in the paracellular transmigration of leukocytes, such as PECAM-1, CD99, and JAM-A, were also found encircling transcellularly migrating leukocytes59,80. Antibodies against these antigens also interfered with the transcellular transmigration, although the role of these antigens in transcellular migration is unknown at present.
Two hallmarks of inflammation are leukocyte extravasation and plasma leaks. Since they are often coinciding, it was debated for a long time whether leukocyte diapedesis would be the cause for the increase in vascular permeability. Arguments against this are based on cases where both events were documented at different sites in the vascular bed of inflamed tissues81–84 and at different times during the inflammatory process85,86. Thus, mechanisms must be in place to ensure a tight endothelial barrier, although leukocytes open endothelial junctions and transmigrate through them. Recently, it was reported that such a mechanism is based on ICAM-1-triggered activation of RhoA, mediated by the RHO GEFs Ect2 and LARG (Figure 1B). This stimulates the activation of the kinase ROCK2b, which in turn activates acto-myosin-based endothelial pore confinement87.
Interestingly, this study showed that interfering with endothelial RhoA in vitro and in vivo caused neutrophil-induced vascular leaks but did not inhibit the transmigration of neutrophils and this is in agreement with other in vitro studies58. Thus, RhoA activity in endothelial cells is redundant for leukocyte transmigration, which is in contrast to other reports discussed above, which suggests RhoA triggered actomyosin pulling on endothelial junctions as a facilitator of the diapedesis process21,76. Although this is clearly a novel view of the role of RhoA in the diapedesis process, it does not argue against the concept of radial actomyosin stress fibers as being responsible for opening junctions by exerting pulling forces. It is possible that other GTPases are responsible for the activation of non-muscle myosin II. Furthermore, calcium/calmodulin is able to activate MLCK; thus, leukocyte-induced activation of non-muscle myosin II would not necessarily require RhoA.
A more dramatic type of leak formation that is linked to the extravasation of neutrophils is visible only under thrombocytopenic conditions (that is, when platelets are depleted). It was shown that, under such conditions, local bleedings (petechiae) occur at sites of local inflammation88. Recently, it was shown that it is the diapedesis of neutrophils which triggers the exit of erythrocytes under conditions of thrombocytopenia54. This implies that platelets prevent the exit of erythrocytes at sites of neutrophil extravasation. Mechanistically, it was shown that platelets require the ITAM receptors CLEC2 and GPVI89 for this protective effect against erythrocyte leaks and it was suggested that single platelets seal neutrophil-induced vascular breaches90.
Recent years have provided the first mechanistic insights into the process of leukocyte diapedesis through the endothelial barrier. Although several cell surface adhesion receptors have been identified at endothelial cell contacts that are involved in the transmigration process (and not in leukocyte capturing and docking), the first signaling mechanisms triggered by PECAM-1 and by CD99 were identified only recently. It will be a major goal in the future to elucidate the potential roles of the various receptors for the following functions: signaling to leukocytes to convey the information that an exit site has been reached; opening and possibly sealing of gaps in the endothelial barrier; participation in leukocyte migration; and facilitating mechanisms that enable leukocytes to overcome the basement membrane. The last step of migration through the basement membrane is still enigmatic91–93. It is an interesting concept that the composition of the basement membrane and low expression regions of certain components of the basement membrane represent preferred sites of transmigration94,95, which may even have an impact on the ability of associated endothelial cells to serve as preferred entry sites. Likewise, pericytes could have guiding effects in this respect96. Finally, it was recently discovered that the transmigration process is not always unidirectional and that under certain conditions neutrophils can also revert the direction and move back into the circulation34,97,98. It will be interesting to elucidate the physiological and pathophysiological relevance of this process.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)