Keywords
H5N8, Guangdong, hemagglutinin, neuraminidase, reassortment, phylogenetics
H5N8, Guangdong, hemagglutinin, neuraminidase, reassortment, phylogenetics
The H5N8 subtype of influenza A virus was first isolated in Ireland in 19831. Until the 2014 outbreak in Korea this was a relatively rare subtype that only occurred sporadically. This Korean outbreak was more sustained and over a much wider geographical region than the earlier outbreaks. The Korean outbreak was also important because it resulted from a viral reassortment which contains the highly pathogenic H5 segment originally isolated from a goose in Guangdong in 19962. The epidemiology of the Guangdong H5 containing H5N8 virus has been investigated extensively3–7 but there is much less investigation of the subtypes that contain the non-Guangdong H5 which has been the predominant hemagglutinin in North America. The most significant finding from these previous studies is that the H5 Guangdong hemagglutinin has now been introduced to North America via bird migratory pathways and that this H5 has not undergone subsequent reassortment into other avian influenza subtypes including H5N2.
An interesting question is why there are large gaps in the history of the sampling of the H5N8 subtype? This is particularly true in the case of the non-Guangdong H5 containing sequences. In order to create a complete history of the H5N8 subtype a phylogenetic analysis of all of the H5 hemagglutinin and N8 neuraminidase sequences was undertaken.
There are three possible explanations for the breaks in detection. They could result from inadequate sampling of the H5N8 avian influenza, the virus might have been present in wild birds, but because avian influenza is often asymptomatic it might be only cryptically expressed, or H5N8 might occur sporadically because it has been created by reassortment events but the new reasserted virus does not spread widely because it is not competitive with alternative reassortment subtypes and so it does not form a continuous population.
A systematic environmental study of bird diseases in the Delaware Bay, as part of the Southeastern Cooperative Wildlife Disease Study8 has only reported the H5N8 subtype sporadically providing evidence against inadequate sampling and cryptic expression. With a systematic collection of biological and environmental samples if a sustained viral population had been present at this location then it is likely that it would have been detected even if infection is asymptomatic.
This leaves the alternative hypothesis that the H5N8 virus occurs sporadically as a result of reassortment events, but that these events do not produce a sustainable H5N8 viral population. This hypothesis can be tested by constructing the complete phylogenetic trees of H5 and N8. The H5N8 subtype samples that fall within a single clade of the H5 hemagglutinin and N8 neuraminidase phylogenetic trees will most likely be the product of a single reassortment. If the H5N8 subtype sequences are scattered widely across the phylogenetic trees, then this would indicate multiple reassortment events that have generated the H5N8 subtype from other subtypes.
This paper shows that the H5N8 subtype is distributed widely across both the H5 and N8 phylogenetic trees and that the sporadic nature of H5N8 is a result of multiple reassortment events that have generated the subtype rather than cryptic expression of the virus.
All of the available H5 hemagglutinin segments (4007 sequences) and N8 neuraminidase segments (1840 sequences) were downloaded from the NCBI Influenza Virus Resource on the 27th of June 20159. The search was restricted to full-length sequences from any host. Manual inspection and editing of the sequences was carried out using Mega6.0610. During manual editing the 5' end of the sequence was edited to remove the un-translated region. All sequences were trimmed to the start codon and stop codons. Sequences with missing nucleotides were removed.
The H5 clades for the H5N8 subset of hemagglutinin sequences were assigned using the Highly Pathogenic H5N1 Clade Classification tool available as part of the Influenza Research Database11–13. While this tool was created for the H5N1 subtype, the recent H5N8 outbreak has been identified as belonging to the new 2.3.4.4 subclade that is part of the classifier.
The H5 hemagglutinin and N8 neuraminidase sequences were aligned using Muscle v 3.8.31. FastTree2.1 was used to create a maximum likelihood tree for all of the sequences using the GTR + gamma evolutionary model14.
Fasttree -boot 10000 –nt –gtr –gamma –quote filename.fas > filename.tree
Given the large number of taxons it is computationally challenging to calculate non-parametric boot-strapped trees, instead FastTree calculates a local support values of each of the splits within the tree using the Shimodaira-Hasegawa (SH) log likelihood test15. This has been shown to have a high correlation to non-parametric bootstrap values14.
The resulting trees were edited, visualised and annotated with FigTree 1.4.216. The trees were displayed as phylograms in order to examine the effect of sampling. The full name, chronological and geographical information was included in the trees as these are essential for determining the homogeneity of the clades. Nodes were labelled with the support values calculated by FastTree, which are a Log Likelihood Ratio. Trees and sub-trees were all rooted to the earliest chronological sequences within the tree.
Supplementary data-files for the phylogenetic analysis of the H5 hemagglutinin are available from http://dx.doi.org/10.5281/zenodo.20653 and for the N8 neuraminidase from http://dx.doi.org/10.5281/zenodo.20655.
Table 1 shows a summary of the H5N8 sequences that are not classified as being members of the 2.3.4.4 clade by the influenza database pathogenic H5N1 classification tool. The complete table of results is given in supplementary table 1. All of the sequences from the Korean outbreak are classified as part of the 2.3.4.4 clade and this also includes many of the 2014 North American sequences but not Californian quail sequence KP101004, which is part of the American non-Guangdong classification. There are two non-Guangdong clades that can be identified as sources of H5 hemagglutinin in H5N8, an American and Eurasian clade.
The Quang Ninh sequence belongs to the Guangdong grouping but it is part of a different sub-clade, 2.3.2.1c. This subclade contains H5N1 sequences that were found in long range migratory birds such as Geese, Cranes and Whooper Swans in Mongolia and Japan between 2009 and 2011. This suggests that this sequence occurred from a distinct reassortment in migratory birds to that which produced the Korean outbreak.
These results show that the H5N8 hemagglutinins are widely distributed across the H5 clades and that almost all of the North American sequences fall outside of the clades within the current nomenclature system12. This demonstrates that there have to have been multiple reassortment events between different H5 and N8 clades which have generated the H5N8 subtype. These need to be explored through a more detailed analysis of the complete phylogenetic trees of the H5 hemagglutinin and N8 neuraminidase. Four distinct reassortment events are already clear. One involving the 2.3.4.4 Guangdong clade, another involving the 2.3.2.1c Guangdong clade and two more involving the Eurasian and American non-Guangdong H5 hemagglutinins.
The full H5 tree contains 4007 sequences, and is rooted on the 1959 Scottish H5N1 hemagglutinin sequence CY015081 (Figure 1). The tree has been collapsed into two main clades which correspond to American (clade 1) and Eurasian sequences (clade 2). There is then a small cluster of ancestral sequences to these groups that includes the Irish H5N8 sequences (shown in red) that form a subclade with an H5N2 sequence from Italy in 1980 and the German H5N6 and H5N2 sequences from 1984–1985. This Irish group represents the first recorded reassortment that produced the H5N8 subtype. Given that its sequence neighbours both before 1983 and afterward are from the H5N2 subtype it is plausible that the H5N8 hemagglutinin originated in the H5N2 subtype. Within the highly pathogenic avian influenza H5N1 classification the Irish H5 sequences are attributed to the American non-Guangdong clade but this more detailed analysis show that there is a European ancestral group that predates the American clade and that the Irish sequences belong there.
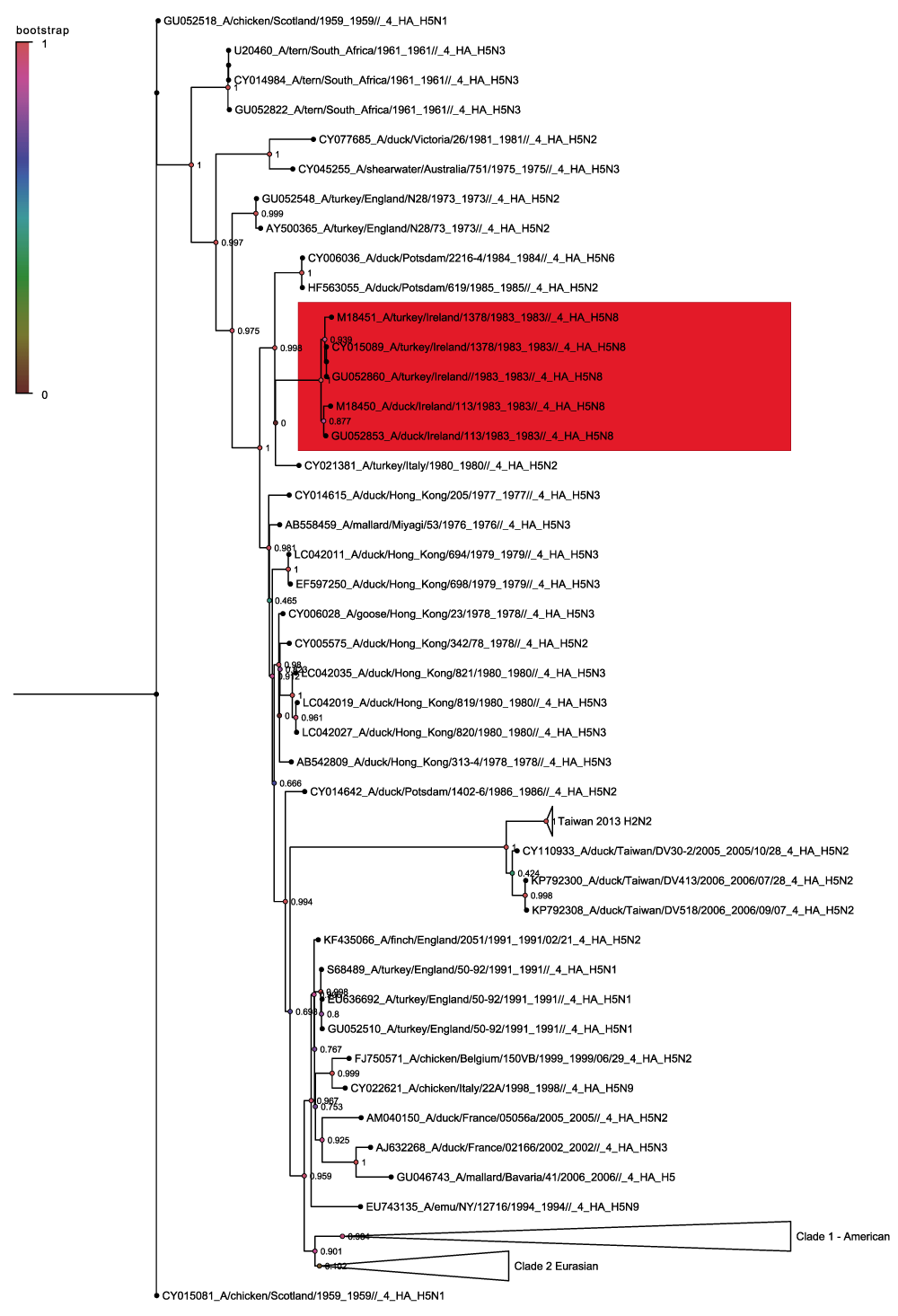
Nodes are labelled and coloured with the local bootstrap likelihood values. The H5N8 sequences are highlighted in red.
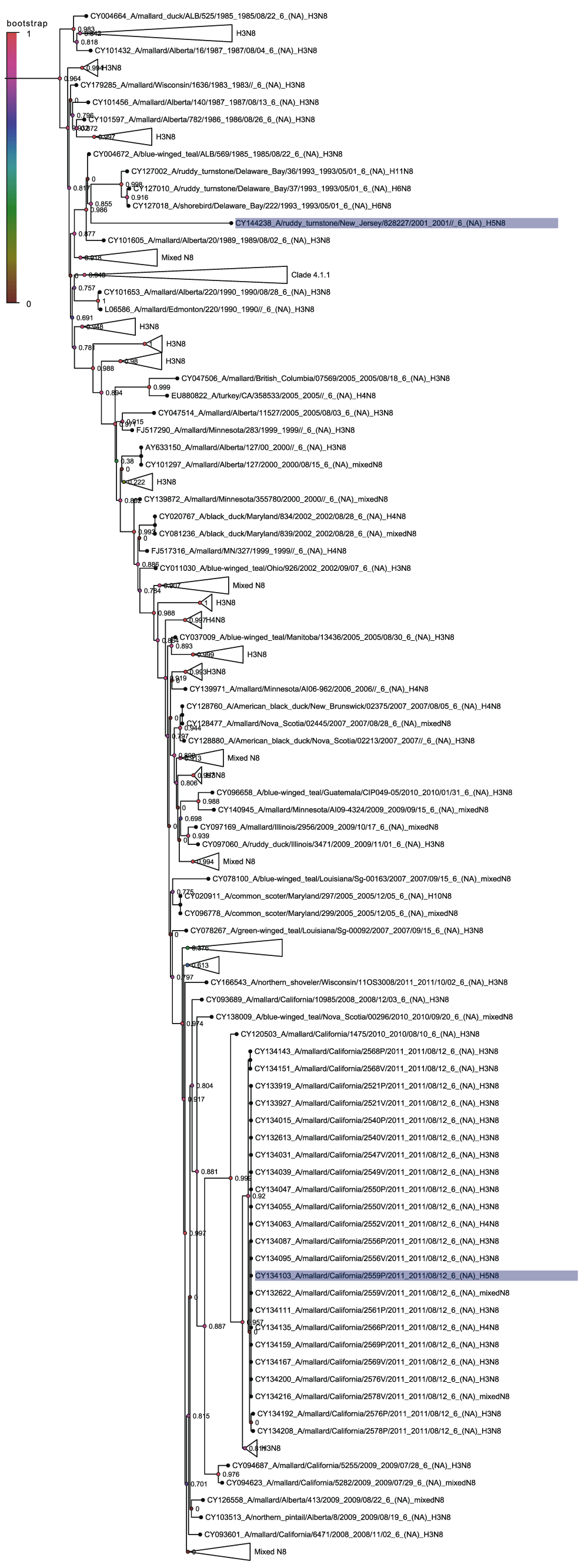
Figure 2 shows clade 1, the American non-Guangdong clade, rooted on the 1966 H5N9 sequence from a Turkey in Ontario (AB558456). This clade contains the H5N8 sequences from Colorado 2006, a quail in California in 2014, a mallard in California in 2011 and a ruddy turnstone in New Jersey in 2001. Each of these appears as a single sequence in a subclade made up of other non-H5N8 viral subtypes. This suggests that each of these occurrences of H5N8 is the result of a different reassortment event.

Nodes are labelled and coloured with the local bootstrap likelihood values. The H5N8 sequences are highlighted in blue.
The 2001 New Jersey sequence is in a subclade with H5N2 and H5N7 virus sequences collected in the same location in the same year. However, it is not clear which subtype is the source of the H5 hemagglutinin. The 2006 Colorado sequence is part of a subclade with the H5N2 subtype and there are a number of other H5N2 viruses from Arkansas, Minnesota and Wisconsin that are closely related and that were also detected in 2006. It is therefore most likely that a reassortment took place between H5N8 and H5N2. The 2011 California sequence forms a distinct subclade where the other members are all H5N1 sequences also from California and also from 2011. This suggests that the H5N8 is the result of a reassortment between H5N8 and H5N1. The 2011 Californian sequence, 2006 Colorado and California 2014 sequences are the most similar to each other but the clade is dominated by H5 hemagglutinins from the H5N5 subtype.
The quail sequence from California in 2014 is particularly important because this could easily be mistaken as being part of the main outbreak of the Guangdong H5 containing virus which is found in the other American H5N8 sequences from that year17. However, sequence analysis makes it clear that this is not part of that group and that it is part of the American non-Guangdong clade and that this most likely originated in another reassortment of H5N5 or H5N6 with an N8 containing subtype.
Confusion about the sources of outbreaks can affect the measures taken to prevent the spread of the disease. This is especially true in the case of highly pathogenic avian influenza where there is likely to be a significant economic impact if the outbreak cannot be managed successfully. Both the Japanese and European outbreaks were contained and while the highly pathogenic Guangdong H5 was not allowed to spread widely in domestic flocks it was present in wild birds18–20.
Clade 2 is predominantly Eurasian was rooted on the 1991 H5N3 Altai sequence and can be divided into two subclades (Figure 3). One that contains non-Guangdong Eurasian sequences and a second that contains the Guangdong sequences. The non-Guangdong Eurasian sequences include a single H5N8 virus from a duck in Thailand in 2002, which is classified as Eurasian Non-Guangdong under the existing nomenclature12 (Figure 4). There is a considerable distance between this sequence and any other H5N8 subtype sequences. This is a clear indication that there must have been a reassortment in Thailand to produce the H5N8 and that it was most likely between an H5N2 subtype hemagglutinin and an N8 containing subtype. This new tree shows that the existing nomenclature for H5 sequences outside of the Guangdong lineage does not adequately cover the diversity of this group of sequences.

This tree is rooted on the EU564116 sequence from a duck in Altai in 1991. Nodes are labelled and coloured with the local bootstrap likelihood values. There are both non-Guangdong and Guangdong subclades.
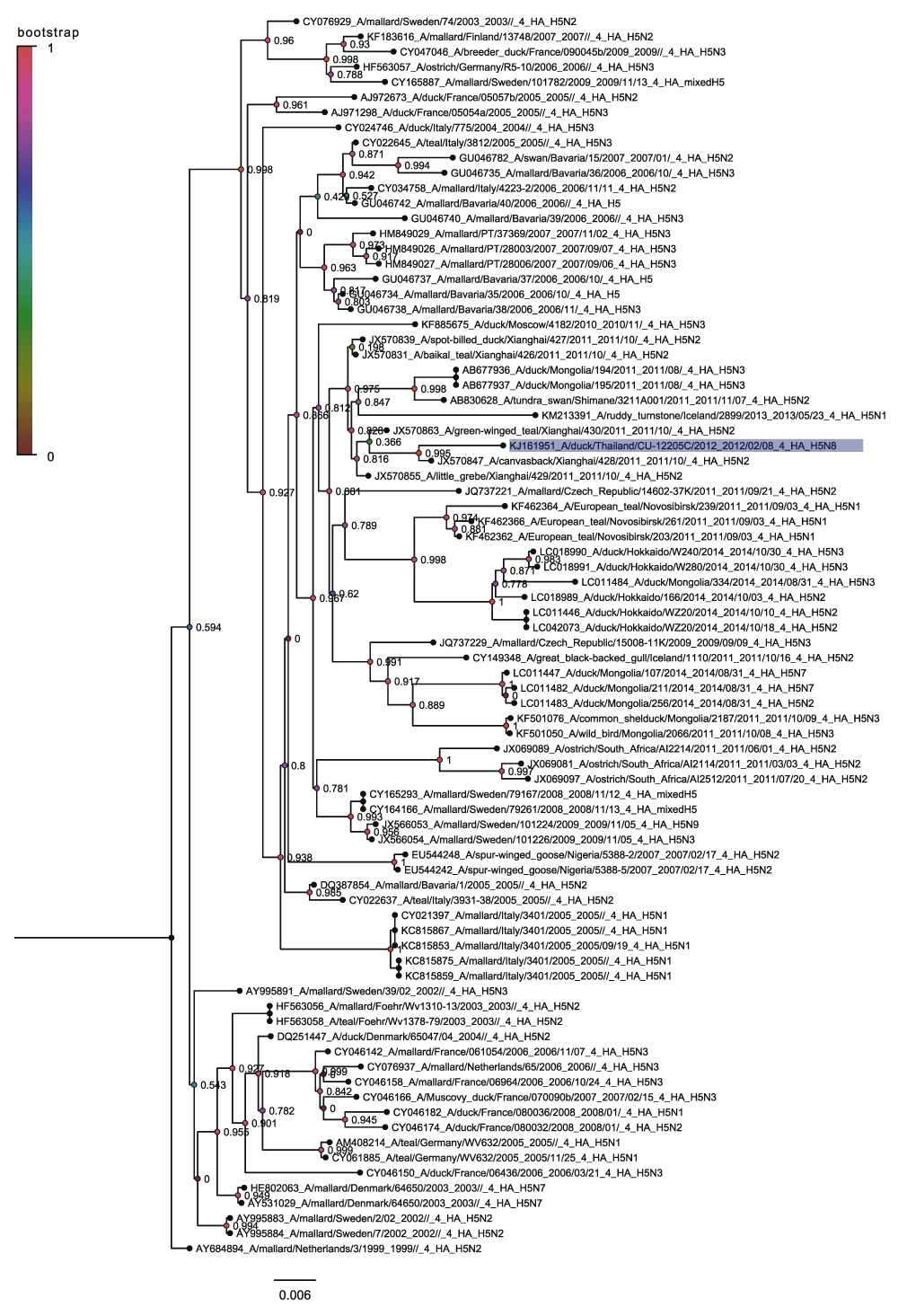
Nodes are labelled and coloured with the local bootstrap likelihood values. The H5N8 sequences are highlighted in blue.
The Guangdong containing subclade is the lineage that has been studied most extensively and this is taken as the prevailing form of pathogenic H5 containing highly pathogenic avian influenza19,21. Within the Guangdong sequences there are three distinct sets of H5N8 sequences. The large bulk correspond to the Buan H5N8 sequences (Figure 5) that have been previously described19. This grouping also includes the North American sequences of H5N8 and H5N2 spread by long distance bird migration22. However the Gochang sequences form a distinct grouping along with Chinese sequences from Zheijiang, Shandong and Jiangsu (Figure 6). This strongly suggests that the Gochang and Buan groups are two different reassortment events even though they are both within the 2.3.4.4 highly pathogenic H5 clade12. The other distinct group contains a single H5N8 sequence from Quang Ninh and is in clade 2.3.2.1c. (Figure 7) again this must have been the product of another reassortment event.
These results from the hemagglutinin trees suggest that there have been a minimum of five reassortment events within the American non-Guangdong sequences, another within the Eurasian non-Guangdong sequences and at least two more in the Guangdong clade. This makes a total of at least eight separate reassortment events that have produced H5N8 from other subtypes. Examining the N8 neuraminidase tree can be used to confirm these reassortment events and to show that absences of H5N8 in the chronological record do not result from poor sampling.

Nodes are labelled and coloured with the local bootstrap likelihood values. The H5N8 sequences are highlighted in blue.

Nodes are labelled and coloured with the local bootstrap likelihood values. The H5N8 sequences are highlighted in blue.
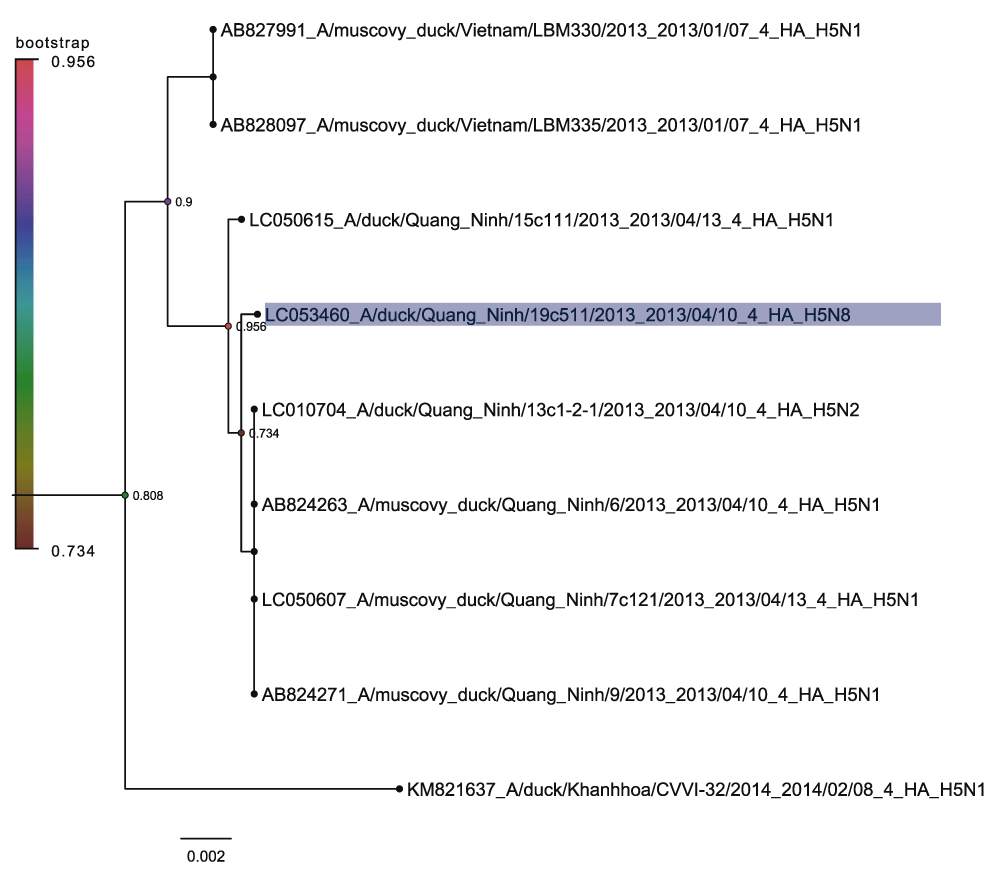
Nodes are labelled and coloured with the local bootstrap likelihood values. The H5N8 sequence is highlighted in blue.
The neuraminidase trees are harder to summarise even though they contain far fewer sequences. This is because they have a less clear clade structure. Figure 8 shows the N8 phylogenetic tree rooted on the 1963 Ukrainian duck sequences. Once again, the H5N8 subtype is close to the root of the tree in the case of the initial Irish outbreak in 1983. There is however, only a single Irish H5N8 sequence amongst a group of H3N8 sequences. Unlike the H5 hemagglutinin there is no existing clade nomenclature for the N8 neuraminidase sequences.
From the neuraminidase tree there are four distinct clades but these are much more heterogeneous than in the case of the H5 trees. The four clades correspond roughly to long range migratory birds, far eastern migratory birds, Gochang and Buan and finally an American clade. Only the Gochang and Buan and the American clade contain the H5N8 subtype and will be considered here.
The simplest of these clades to view is the Gochang and Buan clade where the H5N8 subtypes are clustered tightly together in one subclade with only a few non H5N8 sequences (Figure 9). This highly homogeneous clade is very different to the mixtures of subtypes found in the other clades. The structure of this clade suggests that the Gochang and Buan H5N8 viruses originate from a single source of the N8 neuraminidase but that they divided from one another before the Korean outbreak. This is in agreement with the hemagglutinin trees which show that the H5 hemagglutinins for the Buan and Gochang groups have two more distinct origins. This shows that reassortment can occur on a local level between closely related sequences and produce multiple lineages in the same geographical location.
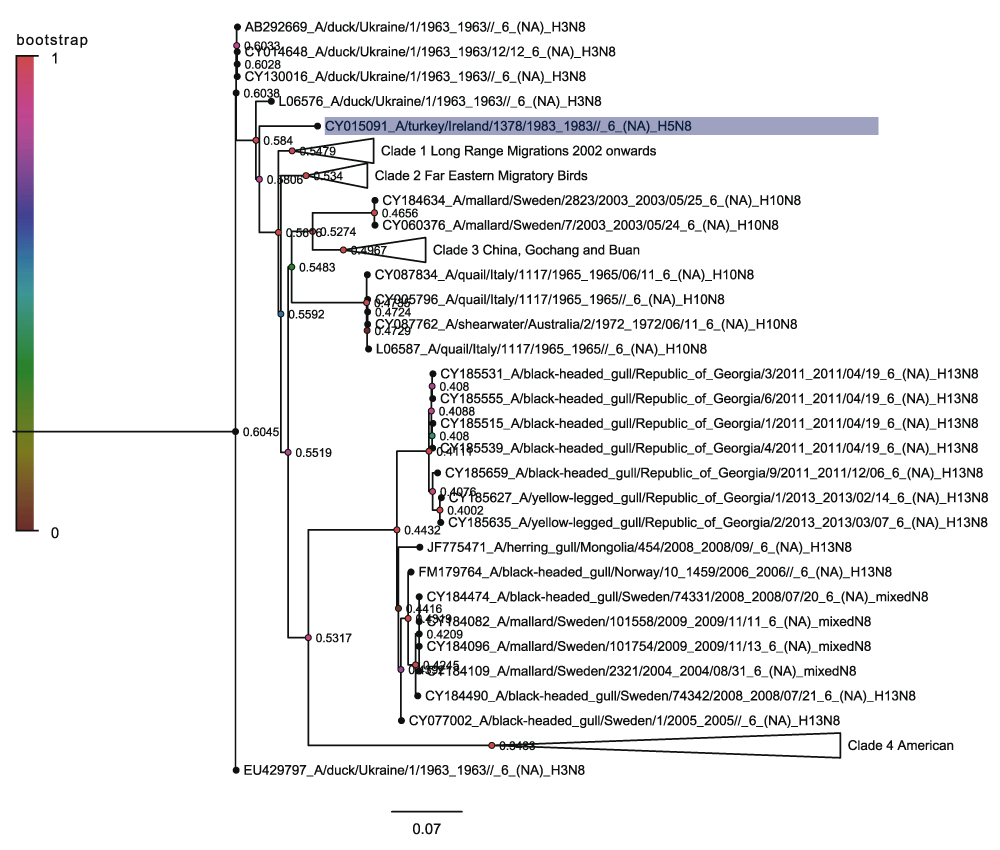
Nodes are labelled and coloured with the local bootstrap likelihood values. The H5N8 sequence is highlighted in blue.

The tree is rooted on the EU429700 sequence from a duck in Eastern China in 2004. The Buan sequences are highlighted in orange and the Gochang sequences are highlighted in blue. Nodes are labelled and coloured with the local bootstrap likelihood values.
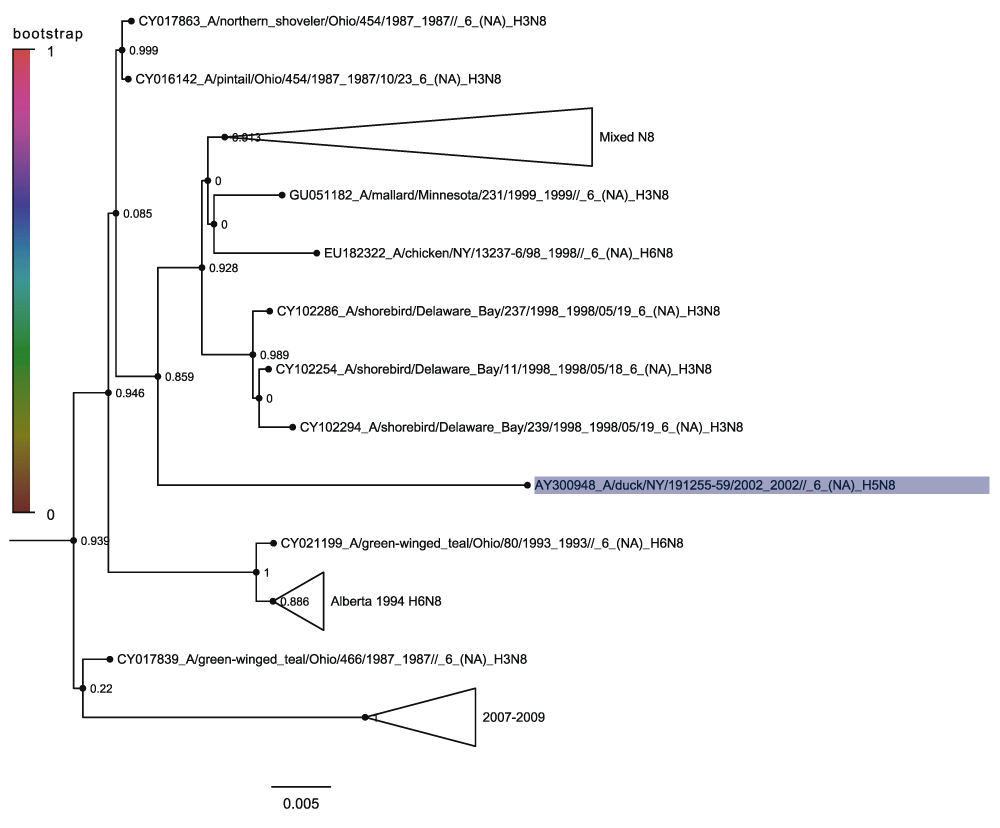
Clade 4 (Figure 10) contains the seven distinct H5N8 sequences that are distributed widely across the clade as singletons. None of the sequences are adjacent to each other in the tree and most are in distantly related subclades. Clade 4.1 (Figure 11) contains the 2001 New Jersey and 2011 Californian H5N8 sequences along with the 2006 Colorado, 2014 California and 2013 Quang Ninh sequences within sub-sub-clade 4.1.1. This agrees with the results of the H5 hemagglutinin sequences phylogenetic tree and is strong evidence that each of the H5N8 viruses corresponds to a reassortment event. What is more significant is the presence of the Quang Ninh sequence amongst the North American sequences as this shows that the N8 neuraminidase circulates more widely than the different H5 hemagglutinin lineages. Clade 4.2 contains the 2002 New York sequence (Figure 13) and clade 4.3 contains the 2012 Thailand sequence (Figure 14). Again this demonstrates how there is a global dispersion of the N8 neuraminidase clade 4, compared to the Guangdong H5 which until 2014 was not present in North America.

This is a collapsed view. Nodes are labelled and coloured with the local bootstrap likelihood values.
Nodes are labelled and coloured with the local bootstrap likelihood values. The H5N8 sequences are highlighted in blue.

Nodes are labelled and coloured with the local bootstrap likelihood values. The H5N8 sequences are highlighted in blue.
Nodes are labelled and coloured with the local bootstrap likelihood values. The H5N8 sequence is highlighted in blue.

Nodes are labelled and coloured with the local bootstrap likelihood values. The H5N8 sequence is highlighted in blue.
The results presented here show that there is a high degree of reassortment that generates new influenza subtypes. The presence of a high proportion of singleton sequences shows that most of H5N8 is often not the preferred subtype as it did not produce a wide-spread outbreak. The Guangdong H5 containing reassortment has produced an H5N8 capable of wider circulation. However it is still possible that once again H5N8 will die out only to return sporadically given that the virus has already undergone further reassortment in North America to produce an H5N2 Guangdong H5 containing subtype23.
In a wider context, reassortment events that create new subtypes need to be accounted for before reliable phylogenetic analysis can be carried out. Sampling for tree construction based on viral subtype, without any consideration of reassortment will be misleading. If we ignore these reassortment events, then we will introduce sampling bias to the trees. Sampling bias is introduced because you selectively sample sequences within a clade that share the same subtype whereas many of the neighbouring hemagglutinin and neuraminidase sequences may actually be from other subtypes.
Where phylogenetic analysis focuses on a viral segment of a specific subtype a complete analysis of all of the sequences for from all subtypes for that segment, as has been performed here, is rarely carried out. The criteria for including sequences in these analyses are usually based on chronological or geographical limits, but these limits reduce the generalizability of the hypothesis being considered. To make sure that sampling is effective a complete phylogenetic analysis of that segment is required. After this then clades and subclades can be selected for further analysis using geographical or chronological criteria. In this way the only bias introduced is that from sequence collection and availability.
This analysis only considered reassortment from the perspective of the glyco-proteins as the reassortment of these protein produces a novel influenza sub-type. Further analysis needs to also include the other viral segments in order to provide a more complete picture of reassortment in avian influenza.
ZENODO: Phylogenetic Analysis of the Influenza H5 hemagglutinins, doi: 10.5281/zenodo.2065324
ZENODO: Phylogenetic analysis of the influenza N8 neuraminidases, doi: 10.5281/zenodo.2065525
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
There are multiple lineages of hemagglutinin in H5N1 and all of the H5N1 ... Continue reading The argument presented in this paper has been proven regardless of the objections of reviewer 2.
There are multiple lineages of hemagglutinin in H5N1 and all of the H5N1 subtypes did not originate from a single H5N1 reassortment event. That was all the paper was saying, nothing about "discovering" reassortment.
The significance of this is that if you create phylogenetic trees of hemagglutinin or neuraminidase that only include sequences from a single subtype you will be missing important data and sequences that form part of their evolutionary history, as shown here.
There are multiple lineages of hemagglutinin in H5N1 and all of the H5N1 subtypes did not originate from a single H5N1 reassortment event. That was all the paper was saying, nothing about "discovering" reassortment.
The significance of this is that if you create phylogenetic trees of hemagglutinin or neuraminidase that only include sequences from a single subtype you will be missing important data and sequences that form part of their evolutionary history, as shown here.