Reddy S and Comai L. Recent advances in understanding the role of lamins in health and disease [version 1; peer review: 2 approved]. F1000Research 2016, 5(F1000 Faculty Rev):2536 (https://doi.org/10.12688/f1000research.9260.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
1Department of Biochemistry and Molecular Biology, Institute for Genetic Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA, USA 2Department of Molecular Microbiology and Immunology, Institute for Genetic Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA, USA
OPEN PEER REVIEW
REVIEWER STATUS
Abstract
Lamins are major components of the nuclear lamina, a network of proteins that supports the nuclear envelope in metazoan cells. Over the past decade, biochemical studies have provided support for the view that lamins are not passive bystanders providing mechanical stability to the nucleus but play an active role in the organization of the genome and the function of fundamental nuclear processes. It has also become apparent that lamins are critical for human health, as a large number of mutations identified in the gene that encodes for A-type lamins are associated with tissue-specific and systemic genetic diseases, including the accelerated aging disorder known as Hutchinson-Gilford progeria syndrome. Recent years have witnessed great advances in our understanding of the role of lamins in the nucleus and the functional consequences of disease-associated A-type lamin mutations. Many of these findings have been presented in comprehensive reviews. In this mini-review, we discuss recent breakthroughs in the role of lamins in health and disease and what lies ahead in lamin research.
Keywords
lamins, nuclear envelope, Hutchinson-Gilford progeria syndrome, Lamin Association Domains,
Corresponding author:
Lucio Comai
Competing interests:
The authors declare that they have no competing interests.
Grant information:
Work in our labs is supported by the National Institute of Neurological Disorders and Stroke and the National Institute of Aging of the National Institutes of Health.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
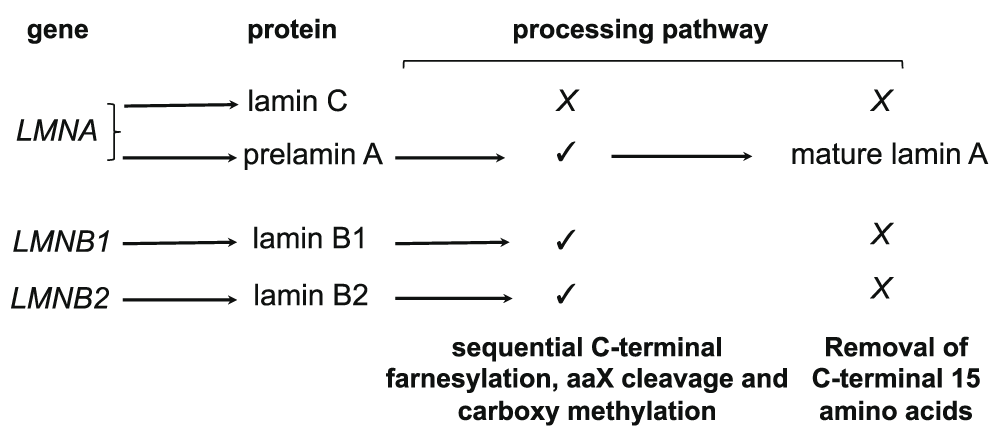
Lamins are members of the family of intermediate filaments that are largely but not exclusively localized to the nuclear lamina, a multiprotein mesh structure found on the inner side of the nuclear membrane of most metazoan cells1–4. Mammalian cells have two types of lamins: A-type lamins, which are expressed in most terminally differentiated cells, and B-type lamins, which are expressed in most or all somatic cells (Figure 1). A-type lamin A and C are encoded by the LMNA gene and generated by alternative splicing, whereas B-type lamin B1 and B2 are encoded by two separate genes: LMNB1 and LMNB2. Short lamin C2 and lamin B3 isoforms encoded by the LMNA and LMNB2, respectively, are expressed only in gametes5,6. Two minor isoforms of lamin A (Δ10) and lamin C (C2) have also been identified, but their function and regulation are not yet fully understood3. Lamin A, B1, and B2, but not lamin C, have a carboxy-terminal CaaX motif (C is cysteine, a is an aliphatic amino acid, and X is any amino acid) that undergoes sequential cysteine farnesylation, aaX cleavage, and carboxy methylation. Whereas these modifications are permanent on lamin B1 and B2, lamin A is synthesized as a prelamin A precursor that undergoes an additional processing step catalyzed by the Zn metallopeptidase STE24 (ZMPSTE24) that removes the carboxy-terminal 15-amino-acid tail, including the modified cysteine to generate mature lamin A. Farnesylation is thought to strengthen the association of B-type lamins with the inner nuclear membrane, while the lack of this modification in lamin A and C allows these lamins to be more loosely associated with the nuclear envelope and also occupy the nucleoplasmic space. Lamins are believed to provide a framework that supports the assembly and stability of the nuclear envelope and contributes to nuclear shape and mechanotransduction1,2,7,8. Moreover, a growing body of research has provided compelling evidence that lamins make significant contributions to the dynamic organization and function of the genome1–4,9. Determining the function of lamins is of critical importance for human health because of the large number of mutations identified across the LMNA gene that are associated with a class of human disorders, collectively known as laminopathies, whose clinical symptoms include skeletal or cardiac muscular dystrophy, lipodystrophy, dysplasia, dermopathy, neuropathy, leukodystrophy, and accelerated aging9,10. The discovery in 2003 that Hutchinson-Gilford progeria syndrome (HGPS), a rare premature aging disease that affects children, is caused by a de novo LMNA mutation that leads to impaired processing of prelamin A and the production of a permanently farnesylated mutant lamin A protein termed progerin11,12 has led to an escalation in lamin research with the hope of finding a cure for this devastating disease. Expression of progerin causes severe cellular defects that affect nuclear morphology, chromatin organization, telomere length homeostasis, DNA repair, nucleoplasmic transport, and redox homeostasis13–17. Recent studies have provided critical information on the contribution of lamins to nuclear mechanics and the spatial organization of the nucleus (Figure 2) and provided considerable experimental evidence for the hypothesis that lamin A mutations disrupt processes that are critical for nucleocytoplasmic mechanotransduction, nuclear positioning, chromatin organization and function, and responses to stress.
Figure 1. Major A-type and B-type lamins in mammals.
Prelamin A, lamin B1, and lamin B2 contain a carboxy-terminal CaaX motif (CSIM in human prelamin A, CAIM in lamin B1, and CYVM in lamin B2; C is cysteine, S is serine, I is isoleucine, M is methionine, A is alanine, Y is tyrosine, and V is valine) which is modified by farnesylation. This is followed by proteolysis of the aaX residues and carboxy methylation at the C-terminal end of lamin A, B1, and B2. Prelamin A undergoes further processing to remove the carboxy-terminal 15 amino acids, including the farnesylated and carboxy methylated cysteine to generate mature lamin A. In Hutchinson-Gilford progeria syndrome cells, the second cleavage site in prelamin A is deleted, and this results in the accumulation of a permanently farnesylated and carboxy methylated prelamin A variant termed progerin. Terminal cleavage of prelamin A is catalyzed by the zinc metallopeptidase ZMPSTE24, an enzyme that has recently been implicated in clearing proteins through clogged endoplasmic reticulum translocon channel98.
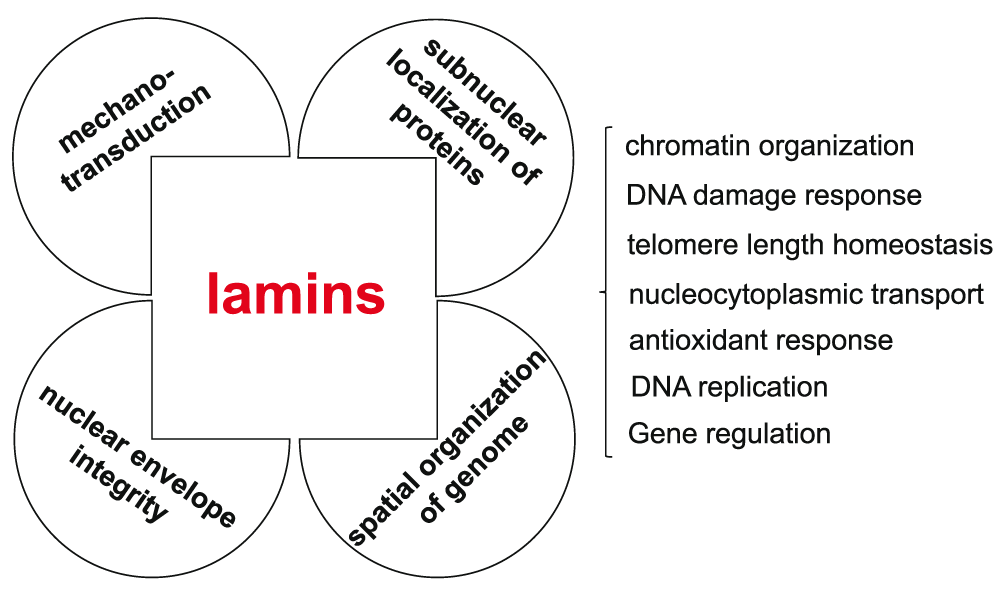
Figure 2. Lamins influence the mechanical properties of the nucleus and contribute to genome organization, function, and stability.
Lamins have roles that support various aspects of nuclear structure and function. Lamins provide mechanical strength to the cell nucleus and contribute to cellular mechanotransduction. Lamins influence the nucleoplasmic environment and contribute to shaping the spatial organization of the genome. Lamins influence genome function and stability by contributing, through interactions with various nuclear factors, to the epigenetic regulation of chromatin, DNA replication and repair, and gene transcription.
Lamins in nuclear mechanobiology
The nucleus plays a critical role in the response to mechanical forces, and new research adds to a growing body of evidence implicating lamin A/C and the linker of nucleo-skeleton to cytoskeleton (LINC) complexes, which bridge the nuclear lamina to the cytoskeleton, in tissue adaptation to mechanical forces7,8,18. Lamins form high-molecular-weight structures, and high-resolution microscopy data have revealed that A- and B-type lamins are organized in a distinct but interdependent meshwork of fibrils19. Each of these structures is likely to contribute to maintaining the organization of the nuclear lamina and the shape of the nucleus. Yet the observation that depletion of lamin A/C increases nuclear deformability in response to mechanical stress suggests that lamin A/C fibrils play a prominent role in regulating the stiffness and elasticity of the nucleus20,21. Consistent with these data, differences in lamin A/C expression leading to changes in lamin A/C-to-B ratio have been demonstrated across distinct cell types, with higher lamin A/C levels observed in cells of tissues often subjected to mechanical torsion, including muscle and heart22. Variations in the lamin A/C-to-lamin B ratio have also been observed during hematopoiesis23, and it is likely that changes in lamin A/C expression affect nuclear stiffness in cancer cells, which may contribute to pathological outcomes, including metastasis24. A recent study has also identified force-dependent changes in lamin A/C conformation25, suggesting that other mechanisms of lamin A regulation contribute to adjusting nuclear shape in response to stress. Research on lamin A/C mutations linked to Emery-Dreifuss muscular dystrophy (EDMD) and dilated cardiomyopathy (DCM) further underscores a role of lamin A/C in nuclear mechanics26–28. These studies demonstrated that several disease-causing mutations compromise the stiffness of the nucleus and the integrity of the nuclear envelope, including the nuclear pore complex, in cells of the affected tissues. Remarkably, a recent report showed that muscle structure and function in an animal model of EDMD with tissue-specific alterations in nuclear mechanics are returned to normal by gene inactivation of the enzyme responsible for protein prenylation29. Although the precise mechanism underlying this observation remains to be determined, it is possible that changes in the properties, physical interactions, or high-order structure formed by unfarnesylated lamin B confers protection against tissue-specific mechanical stress in this animal model. It is important to point out that not all LMNA gene mutations linked to EDMD or DMC, nor mutations associated with familial partial lipodystrophy, result in nuclear fragility27,29, suggesting that distinct mechanical properties or nuclear functions are affected by different lamin A mutations.
Lamins in chromatin structure and spatial organization of the genome
Within the past few years, efforts have been directed at better understanding the relationship between lamins and genome organization and stability. Both A- and B-type lamins bind DNA in vitro30 and associate with chromatin in vivo2,31, and their loss affects genome integrity32–34. Analysis of chromatin-lamin interactions using an in vivo tagging approach (DNA adenine methyltransferase identification, or DamID)35,36 demonstrated that lamins make dynamic contacts with large regions of chromatin, which have been termed lamina-associated domains (LADs), adjacent to the nuclear lamina. These domains are enriched in repressive histone markers, including dimethylated H3K9 and trimethylated H3K27, suggesting that LADs represent a repressive chromatin environment. In spite of these findings, the role of lamins in the formation of LAD remains unclear. A recent study has indicated that lamin C is sufficient for LAD formation at the nuclear lamina37, and another has questioned the need of any lamin for the formation of these domains38. Interestingly, whereas the DamID studies suggested a very high degree of concordance between lamin A/C- and lamin B-associated chromosome domains, recent work using a chromatin-immunoprecipitation approach has identified a subpopulation of lamin A/C that interacts with active regions of chromatin, in coordination with the lamin-associated factor LAP2α39. These are likely interactions that occur within the nucleoplasmic space away from the nuclear lamina since LAP2α colocalizes with lamin A/C within the nuclear interior40,41. Importantly, both LAP2α levels and the nucleoplasmic pool of lamin A/C are dramatically reduced in the presence of the lamin A mutant progerin42,43, and these changes are thought to influence processes that are critical for cell proliferation. The conclusion of this and other recent studies on this topic is that a tight balance between lamin A/C and LAP2α must be maintained to ensure proper cell function, although how this is achieved remains to be worked out. Other studies have also demonstrated that lamins, together with other components of the nuclear lamina termed nuclear envelope transmembrane proteins (NETs), contribute to tissue-specific organization of the genome and influence gene expression by securing peripheral heterochromatin to the nuclear lamina and repositioning genes within the nucleus during cell differentiation44,45. The NET lamin B receptor (LBR) has also been recently implicated in the recruitment of the X chromosome to the nuclear lamina to promote X-inactive-specific transcript (Xist)-mediated gene silencing46. Taken together with the observation that muscle-specific chromatin reorganization is disrupted in an animal model of EDMD28, these findings suggest that altered spatial organization of heterochromatin or incorrect positioning of genes contributes to the development of tissue-specific pathologies in at least a subset of the diseases that have been linked to mutations in lamins or NETs.
Lamin A and the mutant progerin have been shown to differentially influence the stability and spatial localization of epigenetic regulators of chromatin structure31,47, and several studies have reported a gradual decrease in peripheral heterochromatin and global loss of several histone markers of heterochromatin in progerin-expressing cells48–52. However, a recent study has added a twist to this story by showing that increased levels of the heterochromatic histone modification trimethyl H3K9 contribute to the development of the progeroid phenotype53. The authors demonstrated a direct interaction between lamin A and SUV39h1, a chromatin modifier that is responsible for H3K9me3. Progerin also binds SUV39h1, albeit more tightly than lamin A, which results in increased levels of H3K9me3 in progeria cells. This is an unanticipated result that differs from other studies. A clarification of the type of epigenetic changes caused by progerin requires further investigation, but it is possible, as suggested by the authors of this study, that the decreased heterochromatinization reported by others reflects an in vitro cell passage-dependent effect rather than an in vivo process. The concept that progerin disrupts lamin A-protein interactions that locally influence chromatin organization is supported by another recent study54. In this work, lamin A is shown to recruit chromatin modifiers through interactions with barrier-to-autointegration factor (BAF), a family of proteins that are thought to mediate interactions between various factors and chromatin55. As seen with SUV39h1, progerin binds stronger than lamin A to BAF and this interaction results in BAF mislocalization, leading to epigenetic changes that alter chromosome organization and are likely to contribute to cell dysfunction.
Lamins in the regulation of nuclear processes
Fundamental nuclear processes such as transcription, replication, and DNA repair are tightly connected to the spatial organization of the genome and their function relies on the timely recruitment of specific factors to the proper chromosome locations. Recent studies have suggested that progerin disrupts these processes by preventing the recruitment of specific factors to their target site. One example is sirtuin 6 (SIRT6), a protein involved in multiple processes related to genomic stability, stress resistance, telomere maintenance, and energy homeostasis56. A study has shown that both lamin A and progerin bind SIRT6, but a stronger interaction with progerin results in SIRT6 sequestration to the nuclear lamina, which prevents SIRT6 from relocalizing to sites of DNA damage. Taken together with prior data showing that progerin affects the function of other DNA repair factors16, these results underscore the significant hurdle imposed by this mutant lamin A on the pathways that maintain genome integrity. Intriguingly, SIRT6 also plays a role in the recruitment to telomeres of the Werner syndrome protein (WRN)57, a protein whose loss-of-function mutations cause genetic instability leading to an adult-onset type of progeria58. Although it is not known whether WRN function is affected in cells expressing progerin, it is possible that mislocalization of SIRT6 prevents WRN recruitment to telomeres, and this may contribute to telomere dysfunction in HGPS cells. Unfortunately, overexpression of SIRT6 is not sufficient to rescue progeria cell dysfunction, thus limiting the usefulness of potential SIRT6-based therapeutic interventions59.
In support of the idea that sequestration by progerin is a major mechanism leading to cell dysfunction, it has recently been reported that progerin binds NRF2, a transcription factor that regulates the expression of genes involved in maintaining redox homeostasis60, and relocates it to the nuclear lamina61. Oxidative stress, which has been linked to defective nucleocytoplasmic transport and is likely contributing to persistent DNA damage in HGPS cells62–66, appears to be a central factor in the pathophysiology of progeria. Since ectopic expression of constitutively active NRF2 ameliorates several of the cellular defects of progeria cells, deregulation of NRF2 function has been suggested to be a primary driver of accelerated aging. Although it is unclear how constitutively active NRF2 escapes sequestration to the nuclear lamina by progerin, these findings suggest that therapeutic approaches that restore NRF2 function may be beneficial to patients with HGPS. Deregulation of NRF2 has also been observed in cells from muscular dystrophy patients expressing certain missense lamin A mutants that tend to mislocalize to the cytoplasm67. However, this study reported activation rather than repression of NRF2 in these cells through a mechanism that does not involve lamin A binding.
Therapeutic approaches to Hutchinson-Gilford progeria syndrome
Translation of basic science findings into therapeutic approaches is the uttermost goal of biomedical research. In this regard, the Progeria Research Foundation (http://www.progeriaresearch.org), a non-profit organization founded by the parents of a child with HGPS, has been influential in raising awareness and funds for research on finding a cure for this disease, and these efforts have contributed significantly to the large increase in lamin A research during the last decade. The cellular toxicity of partially processed prelamin A mutants like progerin is due primarily to the presence of the farnesyl group at the carboxy-terminal cysteine. Drugs that inhibit protein farnesyl transferase (farnesyl transferase inhibitors, or FTIs) have been shown to improve the cellular phenotype of progeria cells and ameliorate the pathology of mouse models of the disease68–79. FTIs may also hold therapeutic potential for patients carrying EDMD-linked mutations29. Driven by these findings, the Progeria Research Foundation sponsored a single-arm clinical trial using the FTI lonafarnib and reported improvements in weight gain, bone structure, and the cardiovascular system of patients with progeria80. However, FTIs are far from being a cure for progeria and better drugs are urgently needed. Since then, a new clinical trial using pharmacological inhibitors of the mevalonate biosynthetic pathway (pravastatin, zoledronic acid, and lonafarnib) has been under way, and preliminary findings have just been published81. They indicate that even though the three-drug regimen improves bone size and mineral density, no additional benefit over the one-drug treatment is observed in cardiovascular structure and function. Small molecules that reduce the accumulation of progerin (that is, rapamycin) or influence the microtubule network (that is, remodelin) have recently been shown to have beneficial effects in tissue culture models of progeria82–84, and they offer new opportunities for therapeutic intervention. Rapamycin may have a therapeutic effect on other laminopathies, since temsirolimus, a rapamycin analog, has been shown to counteract the deterioration of cardiac function in a murine model of cardiomyopathy caused by a lamin A mutation85. Future studies in animal models will be crucial to better understand the efficacy and usefulness of these and other new drugs in treating patients with LMNA mutations.
Future challenges
The number of articles published on lamins has grown exponentially during the last few years, and tremendous progress has been made in understanding the biological properties of these proteins and lamin A mutants associated with disease. In spite of this gained knowledge, a number of challenges remain. More studies are needed to better understand the relative contributions of lamin A and lamin C to the dynamic spatial organization of the genome in different cell types during development and differentiation. The potential role of lamins in organizing transcription or replication units and DNA damage repair foci needs to be further explored, and future investigations are expected to provide important insights on these topics. Relatively little is known about the molecular mechanisms of tissue-specific disorders caused by LMNA missense mutations that do not affect prelamin A processing. A study in cells from a mouse model of DCM has recently shown that expression of a missense mutant N195K-lamin A (N195K) impairs nucleocytoplasmic shuttling of a key factor in cardiac development86. These results suggest that a single amino acid change in the lamin A polypeptide induces structural alterations that influence the intracellular distribution and function of a cell-type-specific factor. In a new report, two missense LMNA mutations linked to muscular dystrophy (R453W and R482W) have been shown to disrupt LAD and alter heterochromatin organization during myogenic differentiation87. These findings strengthen the idea that lamin A/C contributes to the spatial and structural remodeling of chromatin that takes place during cell differentiation. There are hundreds of mutations in the LMNA gene known to be associated with tissue-specific diseases9. Thus, one may speculate that at least some of these mutations cause tissue-specific defects by affecting the localization or subcellular distribution of factors that, by regulating cell-type-specific regulatory genes or pathways, orchestrate the spatial organization and function of the nucleus. There is also more to learn about the functions of lamin B1 and B2, which, in spite of the high degree of sequence conservation, do not seem to be functionally redundant88,89. There is strong evidence that B-type lamins are required for DNA replication, and recent work has identified a specific role for lamin B1 during the elongation phase of this process32,90–92. Both lamin B1 and B2 have also been implicated in neuronal migration and survival, and altered distribution of the nuclear pore complex has been observed in lamin B1-deficient cortical neurons93–97. This defect has been suggested to affect nucleocytoplasmic shuttling of certain factors97, which is reminiscent of the cellular defect caused by the lamin A mutation associated with DCM discussed above86. These are findings that bring excitement as well as challenges to an area of research that is predicted to expand further over the next several years.
The authors declare that they have no competing interests.
Grant information
Work in our labs is supported by the National Institute of Neurological Disorders and Stroke and the National Institute of Aging of the National Institutes of Health.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We apologize to the many authors whose important work could not be cited owing to space constraints. We thank members of the Comai and Reddy labs for valuable suggestions.
Faculty Opinions recommended
References
1.
Holmer L, Worman HJ:
Inner nuclear membrane proteins: functions and targeting.
Cell Mol Life Sci.
2001; 58(12–13): 1741–7. PubMed Abstract
| Publisher Full Text
2.
Goldman RD, Gruenbaum Y, Moir RD, et al.:
Nuclear lamins: building blocks of nuclear architecture.
Genes Dev.
2002; 16(5): 533–47. PubMed Abstract
| Publisher Full Text
3.
Gruenbaum Y, Foisner R:
Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation.
Annu Rev Biochem.
2015; 84: 131–64. PubMed Abstract
| Publisher Full Text
4.
Burke B, Stewart CL:
The nuclear lamins: flexibility in function.
Nat Rev Mol Cell Biol.
2013; 14(1): 13–24. PubMed Abstract
| Publisher Full Text
5.
Furukawa K, Hotta Y:
cDNA cloning of a germ cell specific lamin B3 from mouse spermatocytes and analysis of its function by ectopic expression in somatic cells.
EMBO J.
1993; 12(1): 97–106. PubMed Abstract
| Free Full Text
6.
Furukawa K, Inagaki H, Hotta Y:
Identification and cloning of an mRNA coding for a germ cell-specific A-type lamin in mice.
Exp Cell Res.
1994; 212(2): 426–30. PubMed Abstract
| Publisher Full Text
11.
De Sandre-Giovannoli A, Bernard R, Cau P, et al.:
Lamin a truncation in Hutchinson-Gilford progeria.
Science.
2003; 300(5628): 2055. PubMed Abstract
| Publisher Full Text
14.
Capell BC, Collins FS:
Human laminopathies: nuclei gone genetically awry.
Nat Rev Genet.
2006; 7(12): 940–52. PubMed Abstract
| Publisher Full Text
15.
Ghosh S, Zhou Z:
Genetics of aging, progeria and lamin disorders.
Curr Opin Genet Dev.
2014; 26: 41–6. PubMed Abstract
| Publisher Full Text
16.
Gonzalo S, Kreienkamp R:
DNA repair defects and genome instability in Hutchinson-Gilford Progeria Syndrome.
Curr Opin Cell Biol.
2015; 34: 75–83. PubMed Abstract
| Publisher Full Text
| Free Full Text
20.
Pajerowski JD, Dahl KN, Zhong FL, et al.:
Physical plasticity of the nucleus in stem cell differentiation.
Proc Natl Acad Sci U S A.
2007; 104(40): 15619–24. PubMed Abstract
| Publisher Full Text
| Free Full Text
24.
Bell ES, Lammerding J:
Causes and consequences of nuclear envelope alterations in tumour progression.
Eur J Cell Biol.
2016; pii: S0171-9335(16)30109-1. PubMed Abstract
| Publisher Full Text
26.
Dialynas G, Flannery KM, Zirbel LN, et al.:
LMNA variants cause cytoplasmic distribution of nuclear pore proteins in Drosophila and human muscle.
Hum Mol Genet.
2012; 21(7): 1544–56. PubMed Abstract
| Publisher Full Text
| Free Full Text
28.
Mattout A, Pike BL, Towbin BD, et al.:
An EDMD mutation in C. elegans lamin blocks muscle-specific gene relocation and compromises muscle integrity.
Curr Biol.
2011; 21(19): 1603–14. PubMed Abstract
| Publisher Full Text
29.
Zuela N, Zwerger M, Levin T, et al.:
Impaired mechanical response of an EDMD mutation leads to motility phenotypes that are repaired by loss of prenylation.
J Cell Sci.
2016; 129(29): 1781–91. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
30.
Shoeman RL, Traub P:
The in vitro DNA-binding properties of purified nuclear lamin proteins and vimentin.
J Biol Chem.
1990; 265(16): 9055–61. PubMed Abstract
31.
Han X, Feng X, Rattner JB, et al.:
Tethering by lamin A stabilizes and targets the ING1 tumour suppressor.
Nat Cell Biol.
2008; 10(11): 1333–40. PubMed Abstract
| Publisher Full Text
40.
Naetar N, Korbei B, Kozlov S, et al.:
Loss of nucleoplasmic LAP2alpha-lamin A complexes causes erythroid and epidermal progenitor hyperproliferation.
Nat Cell Biol.
2008; 10(11): 1341–8. PubMed Abstract
| Publisher Full Text
48.
Columbaro M, Capanni C, Mattioli E, et al.:
Rescue of heterochromatin organization in Hutchinson-Gilford progeria by drug treatment.
Cell Mol Life Sci.
2005; 62(22): 2669–78. PubMed Abstract
| Publisher Full Text
| Free Full Text
49.
Goldman RD, Shumaker DK, Erdos MR, et al.:
Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome.
Proc Natl Acad Sci U S A.
2004; 101(24): 8963–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
53.
Liu B, Wang Z, Zhang L, et al.:
Depleting the methyltransferase Suv39h1 improves DNA repair and extends lifespan in a progeria mouse model.
Nat Commun.
2013; 4: 1868. PubMed Abstract
| Publisher Full Text
| Free Full Text
55.
Jamin A, Wiebe MS:
Barrier to Autointegration Factor (BANF1): interwoven roles in nuclear structure, genome integrity, innate immunity, stress responses and progeria.
Curr Opin Cell Biol.
2015; 34: 61–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
58.
Li B, Jog S, Candelario J, et al.:
Altered nuclear functions in progeroid syndromes: a paradigm for aging research.
ScientificWorldJournal.
2009; 9: 1449–62. PubMed Abstract
| Publisher Full Text
| Free Full Text
63.
Richards SA, Muter J, Ritchie P, et al.:
The accumulation of un-repairable DNA damage in laminopathy progeria fibroblasts is caused by ROS generation and is prevented by treatment with N-acetyl cysteine.
Hum Mol Genet.
2011; 20(20): 3997–4004. PubMed Abstract
| Publisher Full Text
64.
Lattanzi G, Marmiroli S, Facchini A, et al.:
Nuclear damages and oxidative stress: new perspectives for laminopathies.
Eur J Histochem.
2012; 56(4): e45. PubMed Abstract
| Publisher Full Text
| Free Full Text
66.
Sieprath T, Darwiche R, De Vos WH:
Lamins as mediators of oxidative stress.
Biochem Biophys Res Commun.
2012; 421(4): 635–9. PubMed Abstract
| Publisher Full Text
68.
Candelario J, Borrego S, Reddy S, et al.:
Accumulation of distinct prelamin A variants in human diploid fibroblasts differentially affects cell homeostasis.
Exp Cell Res.
2011; 317(3): 319–29. PubMed Abstract
| Publisher Full Text
69.
Candelario J, Sudhakar S, Navarro S, et al.:
Perturbation of wild-type lamin A metabolism results in a progeroid phenotype.
Aging Cell.
2008; 7(3): 355–67. PubMed Abstract
| Publisher Full Text
| Free Full Text
70.
Lattanzi G:
Prelamin A-mediated nuclear envelope dynamics in normal and laminopathic cells.
Biochem Soc Trans.
2011; 39(6): 1698–704. PubMed Abstract
| Publisher Full Text
71.
Yang SH, Chang SY, Andres DA, et al.:
Assessing the efficacy of protein farnesyltransferase inhibitors in mouse models of progeria.
J Lipid Res.
2010; 51(2): 400–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
72.
Capell BC, Erdos MR, Madigan JP, et al.:
Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome.
Proc Natl Acad Sci U S A.
2005; 102(36): 12879–84. PubMed Abstract
| Publisher Full Text
| Free Full Text
73.
Capell BC, Olive M, Erdos MR, et al.:
A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a progeria mouse model.
Proc Natl Acad Sci U S A.
2008; 105(41): 15902–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
75.
Glynn MW, Glover TW:
Incomplete processing of mutant lamin A in Hutchinson-Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition.
Hum Mol Genet.
2005; 14(20): 2959–69. PubMed Abstract
| Publisher Full Text
76.
Toth JI, Yang SH, Qiao X, et al.:
Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes.
Proc Natl Acad Sci U S A.
2005; 102(36): 12873–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
77.
Yang SH, Meta M, Qiao X, et al.:
A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation.
J Clin Invest.
2006; 116(8): 2115–21. PubMed Abstract
| Publisher Full Text
| Free Full Text
78.
Mallampalli MP, Huyer G, Bendale P, et al.:
Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson-Gilford progeria syndrome.
Proc Natl Acad Sci U S A.
2005; 102(40): 14416–21. PubMed Abstract
| Publisher Full Text
| Free Full Text
80.
Gordon LB, Kleinman ME, Miller DT, et al.:
Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome.
Proc Natl Acad Sci U S A.
2012; 109(41): 16666–71. PubMed Abstract
| Publisher Full Text
| Free Full Text
82.
Cao K, Graziotto JJ, Blair CD, et al.:
Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells.
Sci Transl Med.
2011; 3(89): 89ra58. PubMed Abstract
| Publisher Full Text
83.
Cenni V, Capanni C, Columbaro M, et al.:
Autophagic degradation of farnesylated prelamin A as a therapeutic approach to lamin-linked progeria.
Eur J Histochem.
2011; 55(4): e36. PubMed Abstract
| Publisher Full Text
| Free Full Text
85.
Choi JC, Muchir A, Wu W, et al.:
Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation.
Sci Transl Med.
2012; 4(144): 144ra102. PubMed Abstract
| Publisher Full Text
| Free Full Text
90.
Moir RD, Montag-Lowy M, Goldman RD:
Dynamic properties of nuclear lamins: lamin B is associated with sites of DNA replication.
J Cell Biol.
1994; 125(6): 1201–12. PubMed Abstract
| Publisher Full Text
| Free Full Text
91.
Moir RD, Spann TP, Herrmann H, et al.:
Disruption of nuclear lamin organization blocks the elongation phase of DNA replication.
J Cell Biol.
2000; 149(6): 1179–92. PubMed Abstract
| Publisher Full Text
| Free Full Text
92.
Spann TP, Moir RD, Goldman AE, et al.:
Disruption of nuclear lamin organization alters the distribution of replication factors and inhibits DNA synthesis.
J Cell Biol.
1997; 136(6): 1201–12. PubMed Abstract
| Publisher Full Text
| Free Full Text
93.
Coffinier C, Chang SY, Nobumori C, et al.:
Abnormal development of the cerebral cortex and cerebellum in the setting of lamin B2 deficiency.
Proc Natl Acad Sci U S A.
2010; 107(11): 5076–81. PubMed Abstract
| Publisher Full Text
| Free Full Text
1
Department of Biochemistry and Molecular Biology, Institute for Genetic Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA, USA 2
Department of Molecular Microbiology and Immunology, Institute for Genetic Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA, USA
Work in our labs is supported by the National Institute of Neurological Disorders and Stroke and the National Institute of Aging of the National Institutes of Health.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Reddy S and Comai L. Recent advances in understanding the role of lamins in health and disease [version 1; peer review: 2 approved]. F1000Research 2016, 5(F1000 Faculty Rev):2536 (https://doi.org/10.12688/f1000research.9260.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
Foisner R. Reviewer Report For: Recent advances in understanding the role of lamins in health and disease [version 1; peer review: 2 approved]. F1000Research 2016, 5(F1000 Faculty Rev):2536 (https://doi.org/10.5256/f1000research.9968.r17102)
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Foisner R. Reviewer Report For: Recent advances in understanding the role of lamins in health and disease [version 1; peer review: 2 approved]. F1000Research 2016, 5(F1000 Faculty Rev):2536 (https://doi.org/10.5256/f1000research.9968.r17102)
Lammerding J. Reviewer Report For: Recent advances in understanding the role of lamins in health and disease [version 1; peer review: 2 approved]. F1000Research 2016, 5(F1000 Faculty Rev):2536 (https://doi.org/10.5256/f1000research.9968.r17101)
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Lammerding J. Reviewer Report For: Recent advances in understanding the role of lamins in health and disease [version 1; peer review: 2 approved]. F1000Research 2016, 5(F1000 Faculty Rev):2536 (https://doi.org/10.5256/f1000research.9968.r17101)
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)