Keywords
RNA-seq, transcriptome, MAKER-P, genome annotation, berberine bridge enzyme, Trinity, walnut genome sequence
RNA-seq, transcriptome, MAKER-P, genome annotation, berberine bridge enzyme, Trinity, walnut genome sequence
The genome of a particular organism is static in all cells, unlike the dynamic transcriptome, which is the transcription of the gene space into RNA molecules in a fashion responsive to a variety of factors, such as developmental stage, tissue, and external stimuli. RNA-seq, a high-throughput RNA sequencing method, has radically transformed the identification of transcripts and quantification of transcriptional levels (Flintoft, 2008; Wang et al., 2009). It is supported by a diverse set of computational methods for analyzing the resulting data (Chakraborty et al., 2015; Chang et al., 2015; Chu et al., 2013; Fu et al., 2012; Grabherr et al., 2011; Lohse et al., 2012; Mbandi et al., 2015; Schulz et al., 2012; Simpson et al., 2009; Trapnell et al., 2009; Trapnell et al., 2012; Wang et al., 2010; Zerbino & Birney, 2008).
Rapid advances in genome sequencing technologies have generated sequences for a deluge of organisms and species. The task of annotating these sequences has been addressed by several flows. These pipelines are categorized in http://omictools.com/genome-annotation-category and http://genometools.org/ and reviewed in (Yandell & Ence, 2012). Here, we focus specifically on MAKER-P (Campbell et al., 2014; Holt & Yandell, 2011; Law et al., 2015; Neale et al., 2014), which was used to annotate the recently published walnut genome sequence (WGS) (Martínez-García et al., 2016).
In the current study, the YeATS suite (Chakraborty et al., 2015) was enhanced to include genome annotation capabilities using RNA-seq-derived transcriptomes (YeATS annotation module - YeATSAM). First, the Trinity-assembled transcriptome obtained from twenty different tissues was compared to the WGS, excluding transcripts emanating from extraneous sources. This step incidentally revealed both biodiversity and plant-microbe interactions in walnut tree(s) from Davis, California (Chakraborty et al., 2016a). The WGS-derived transcripts were split into three open reading frames (ORFs), which were subjected to BLAST analysis using a plant proteome database obtained from the Ensembl database (Kersey et al., 2016). Transcripts can contain more than one significant ORF and must be handled differently depending on whether they map to the same or a different protein. The resulting analysis provided the WGS annotation.
Both MAKER-P and YeATSAM failed to annotate several hundred proteins annotated by the other. Many of the proteins had repetitive sequences or domains that, although difficult to detect, do not represent critical proteins during annotation. An egg cell-secreted protein (Sprunck et al., 2012), a copper chaperone (Shin et al., 2012), and a clavata3/ESR-Related protein (Kinoshita et al., 2007) were among the proteins not detected through the YeATSAM flow. Some proteins undetected in the MAKER-P flow are more significant in the context of a plant genome: several photosynthesis-related proteins encoded by the chloroplast (Nelson & Yocum, 2006) and the large family of FAD-binding berberine bridge enzymes (BBE) involved in biosynthesis of antimicrobial benzophenanthridines (Cheney, 1963; Winkler et al., 2008). We posited possible reasons for such exclusions and recommend incorporating both flows for comprehensive enumeration of genes in the WGS.
As further validation, YeATSAM was applied to chickpea (Cicer arietinum L.), an important pulse crop with many nutritional and health benefits (Jukanti et al., 2012). The RNA-seq-derived transcriptome of chickpea has also been sequenced (Garg et al., 2011) and was processed through the YeATSAM pipeline to identify ~1000 proteins that are encoded by these transcripts, but are not annotated in the NCBI database, most of which were annotated using Gnomon (Souvorov et al., 2010).
The input to YeATSAM is a set of post-assembly transcripts (∅TRS) and the walnut genome sequence (WGS) (Figure 1). Transcripts that do not align to the WGS were removed (Chakraborty et al., 2016a). A BLAST database of protein peptides (plantpep.fasta: 1M seqeunces) using ~30 organisms (list.plants) from the Ensembl genome was created (Kersey et al., 2016). The three longest open reading frames (ORF), obtained using the ‘getorf’ utility in the EMBOSS suite (Rice et al., 2000), for each transcript in (∅TRS) underwent BLAST analysis (Camacho et al., 2013) to the ‘plantpep.fasta’. For cutoff E-value=1E-8, depending on the number of matches, the transcripts were clustered as:
1. None - either a previously unknown gene or non-coding RNA.
2. One - unique ORF.
3. Multiple ORFs matching to the same gene - merge the ORFs if the Evalue of the combined ORF is significantly lower.
4. Multiple ORFs matching to different genes - duplicate the transcripts, associating each transcript with a different ORF.
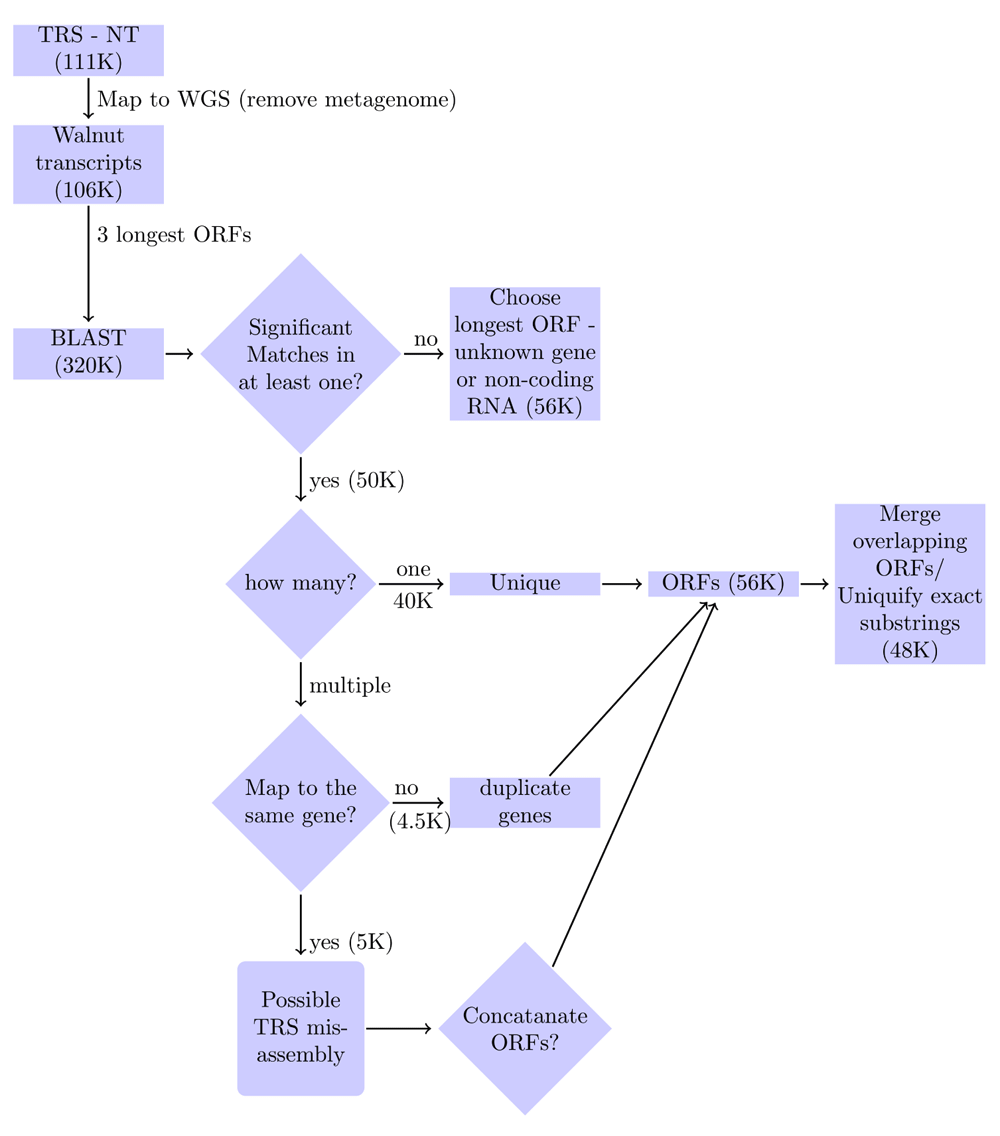
First, transcripts from extraneous organisms are pruned. Next, the three longest open reading frames (ORFs) from each transcript undergo BLAST analysis to a database of plant peptides. Depending on the number of significant matches, the transcripts are clustered as: (a) None - either a previously unknown gene, or non-coding RNA. (b) One - Unique ORF (c) Multiple ORFs matching to the same gene - merge the ORFs if the Evalue of the combined ORF is significantly lower. (d) Multiple ORFs matching to different genes - duplicate the transcripts, associating each with a different ORF. Subsequently, the ORFs are merged based on overlapping amino acid sequences and exact substrings are removed.
Fifteen samples of walnut tissue were gathered from Chandler trees growing in the Stuke block at UC Davis between April and October 2008. Four additional samples were taken from Chandler plant material from the same orchard maintained in tissue culture. Several grams of leaf and root tissue from each plant were frozen in liquid nitrogen and then transferred to a -80 C freezer. RNA was isolated from each sample using the hot borate method (Wilkins & Smart, 1996) followed by purification and DNAse treatment using an RNA/DNA Mini Kit (Qiagen, Valencia, CA) per the manufacturer’s protocol. High-quality RNA was confirmed by running an aliquot of each sample on an Experion Automated Electrophoresis System (Bio-Rad Laboratories, Hercules, CA). The cDNA libraries were constructed following the Illumina mRNA-sequencing sample preparation protocol (Illumina Inc., San Diego, CA). Final elution was performed with 16µL RNase-free water. The quality of each library was determined using a BioRad Experion (BioRad, Hercules, CA). Each library was run as an independent lane on a Genome Analyzer II (Illumina, San Diego, CA) to generate 85bp paired-end sequences from each cDNA library. Over a billion reads were obtained. Prior to assembly, all reads underwent quality control for paired-end reads and trimming using Sickle v1.33 (Joshi & Fass, 2011). The minimum read length was 45bp with a minimum Sanger quality score of 35. The quality-controlled reads were de novo assembled with Trinity v2.0.6 (Grabherr et al., 2011). Standard parameters were used and the minimum contig length was 300bp. Individual assemblies for each library and a combined assembly of all tissues were performed.
The walnut genome sequence has been released to the public domain (http://ucanr.edu/sites/wgig/). The Illumina (Genome Analyzer II) for all 20 tissues can be accessed at http://www.ncbi.nlm.nih.gov/sra/PRJNA232394.
The transcriptome of Cicer arietinum (transHybrid.fasta, ICC4958; Desi chickpea) was obtained from http://www.nipgr.res.in/ctdb.html (Garg et al., 2011). The dataset ‘represents optimized de novo hybrid assembly of 454 and short-read sequence data.’ About two million 454 reads were assembled using Newbler v2.3 followed by hybrid assembly with 53409 transcripts generated by optimized short-read data assembly using TGICL, as reported previously (Garg et al., 2011). The set of annotated proteins from chickpea was obtained from the NCBI database (chickpea.pep.fasta, N=34198).
PHYML v3.0 was used to generate phylogenetic trees from alignments (Guindon et al., 2005). Multiple sequence alignment was done using ClustalW (Larkin et al., 2007) and figures were generated using the ENDscript server 2.0 (Robert & Gouet, 2014). The source code written in Perl is provided as Dataset 1 (YeATSAM.tgz). A README is provided inside the top-level directory for installation and running the programs.
The input to YeATSAM was ~111K Trinity-assembled transcripts (Combined TrinityFull.fasta) (Figure 1). Each transcript was aligned to the WGS (wgs.5d.scafSeq200+.trimmed) using BLAST (Camacho et al., 2013). Transcripts that did not align to the WGS (cutoff BLAST bitscore=75) were excluded (Chakraborty et al., 2016a). Those transcripts that aligned to the WGS (list.transcriptome.clean: 106K) were split into the three longest open reading frames (ORF) (list.transcriptome.clean.ORFS: 320K).
A BLAST database of protein peptides (plantpep.fasta:1M sequences) using ~30 organisms (list.plants) from the Ensembl genome was created (Kersey et al., 2016). The availability of proteomes from related organisms accelerates the annotation. The BLAST results of list.transcriptome.clean.ORFS: 320K on ‘plantpep.fasta’ was processed using a cutoff: bitscore=60, Evalue~=1E-10.
There are two instances in which ORFs can be merged to create a longer amino acid sequence. The first scenario occurs when a particular transcript has multiple ORFs that match to the same protein with high significance, indicating that a sequencing or assembly error has broken a contiguous ORF (Chakraborty et al., 2015). In total, 5% of the present transcripts (5,000 of 106,000) had two or more ORFs matching with high significance to the same protein, exactly mirroring the 5% error rates seen in transcripts restricted to the transcriptome from the tissue at the heartwood/sapwood transition zone in black walnut (Chakraborty et al., 2015). While most of these transcripts have repetitive elements, there were other non-repetitive sequences with this particular problem. C20727_G1_I1 is one example: it has two ORFS, ORF_15 and ORF_36, that match a DNA repair metallo-β-lactamase family protein (Accession number: XP007043420.1) with Evalues=9E-70 and 6E-96, respectively (Figure 2a). The two ORFs were merged (inserting the sequence ‘ZZZ’, although the length of the missing fragment is not known) since the Evalue of the combined ORF reduces to 2E-175 and the merged sequence was chosen as representative for the transcript. ORFs are not merged when the combined ORF did not significantly decrease the Evalue and the longer ORF was selected to represent the transcript.

(a) ORFs from the same transcript: C20727_G1_I1 has two ORFS (ORF 15 and ORF 36) matching to a DNA repair metallo-β-lactamase family protein (Accession number: XP007043420.1) with high significance. We merged the two ORFs (inserting ‘ZZZ’) since the Evalue of the combined ORF is significantly reduced. (b) ORFs from different transcripts: We merged ORFs from two different transcripts (C53209_G8_I1 and C53209_G6_I1), since both transcripts map to the same scaffold (SUPER472) can be overlapped based on the sequence string ‘PNRSSLP’, and the merged ORF has a significantly reduced Evalue.
The other scenario occurs when the assembler fails to merge two transcripts into a single one. In this instance, two ORFs emanating from different transcripts with significant overlaps were merged. While the merging of two ORFs was described previously (Chakraborty et al., 2015), we introduced an additional filter to select mergeable ORFs based on whether the E-value obtained by merging the two ORFs is significantly reduced. For example, transcripts C53209_G8_I1 and C53209_G6_I1 both map to the scaffold SUPER472 and their corresponding ORFs can be merged based on the sequence string ‘PNRSSLP’ (Figure 2b). The individual ORFs and the combined ORFs align to an autophagy-related protein (TAIR ID: AT3G49590.2) with Evalues 2e-106, 8e-63, and 1e-180, respectively. The increased significance of the combined ORF, in addition to other checks, like ensuring that mapping is to the same scaffold, adds further support to the fact that these transcripts should have been contiguous in the final assembled transcriptome.
About 3% of transcripts have ORFs that map to different proteins. Some transcripts should not have been merged. C1089_G1_I1 is an interesting example: a 4574 nt transcript that maps to the chloroplast and encodes two genes. One is highly variable and the other is conserved. The two ORFS, ORF_64 (fwd: 1117-2631) and ORF_108 (fwd: 3195 - 4271), map to maturase K (TAIR ID: ATCG00040.1) and photosystem II reaction center protein (TAIR ID: ATCG00020.1) with very high significance. Maturase K is a good candidate for barcoding angiosperms because it has highly variable coding sequences (Yu et al., 2011), while the photosystem II reaction center protein is completely conserved (100% similarity with Arabidopsis). Another example is C19241_G1_I1 (4702 nt), split into ORF_68 (fwd: 176-3487) and ORF_115 (reverse: 4509-4096) encoding a damaged DNA binding protein (TAIR ID: AT4G05420.1) and photosystem I subunit K (TAIR ID: AT1G30380.1) with high significance, respectively. These transcripts are split in the YeATSAM flow, resulting in one ORF per transcript. Subsequently, this artifact of the Trinity assembly led to several unannotated proteins in the MAKER-P flow.
We compared the annotations of walnut by MAKER-P (walnut.wgs.5d.all.maker.proteins.fasta) and YeATSAM (DB.ORFBEST.60). MAKER-P and YeATSAM each failed to annotate several proteins identified by the other (MAKER-P=~4000; YeATSAM=700). Although most of these unannotated proteins have repetitive sequences (transposable elements), some vital, non-repetitive proteins were excluded by each method. For example, an egg cell-secreted protein (‘WALNUT 00001389-RA’) (Sprunck et al., 2012), a Clavata3/esr-related gene (‘WALNUT 00023705-RA’) (Kinoshita et al., 2007) and a copper chaperone (‘WALNUT 00006344-RA’) (Shin et al., 2012) were not annotated in the YeATSAM flow. These genes do not have transcripts in the twenty tissues analyzed in the current study and are most likely pseudogenes.
MAKER-P fails to annotate many key photosystem-related proteins (Table 1). The transcript C59245_G1_I1 has ORF_43 (fwd: 176-1714) and ORF_70 (fwd: 2212-2496) mapping to photosystem II reaction center protein B (PSBB) and photosystem II reaction center protein H (PSBH), respectively. While MAKER-P does annotate PSBB, it failed to detect PSBH. These proteins map to transcripts encoding two significant ORFs (>1E-10), indicating that failure to handle this might have excluded these proteins. Also, these proteins are encoded by the chloroplast. However, this limitation of MAKER-P is not confined to transcripts emanating from the chloroplast. For example, C48031_G3_I1 encodes a leucine-rich repeat transmembrane protein kinase (AT5G48940.1) and a metallo-β-lactamase family protein (TAIR ID: AT4G33540.1) and is mapped to scaffold ‘SUPER374’. MAKER-P failed to annotate the β-lactamase family protein.
These transcripts have multiple open reading frames (ORFs) mapping to different proteins with high significance. For example, C59245_G1_I1 has another ORF (43) which maps to photosystem II reaction center protein B (PSBB). MAKER-P annotates PSBB, but not PSBH. These transcripts all emanate from the chloroplast, although not all genes that MAKER-P failed to annotate were from the chloroplast. Genes predicted by MAKER-P that are not identified by YeATSAM are listed with their homology to corresponding genes in the TAIR database.
Furthermore, MAKER-P failed to annotate any FAD-binding berberine bridge enzymes (BBE) in the WGS (Kutchan & Dittrich, 1995). These enigmatic enzymes are implicated in the transformation of (S)-reticuline to (S)-scoulerine during benzophenanthridine alkaloid biosynthesis in plants (Winkler et al., 2006). This pathway is over-expressed upon osmotic stress and pathogen attack (Attila et al., 2008; González-Candelas et al., 2010), provides resistance in lettuce, sunflower and transgenic tobacco by generating anti-microbial compounds (Custers et al., 2004), and has unknown functions at specific developmental stages in Arabidopsis (Irshad et al., 2008; Pagnussat et al., 2005). Moreover, it is expressed in floral nectar (Nectarin V, NtBBE) (Carter & Thornburg, 2004) and roots of tobacco (Kajikawa et al., 2011), and in xylem sap of cabbage (Ligat et al., 2011) and grapevine (Chakraborty et al., 2016b). NtBBE was constitutively expressed in the Phytophthora infestans-resistant potato genotype SW93-1015 (Ali et al., 2012). Benzophenanthridines are antimicrobial; the California poppy (Eschscholzia californica) is used as a traditional medicine (Cheney, 1963; Oldham et al., 2010). Oral administration of the alkaloid berberine isolated from a Chinese herb lowered cholesterol in 32 hypercholesterolemic patients over three months (Kong et al., 2004). Berberine has also been shown to possess antidiabetic properties (Lee et al., 2006).
The number of BBE genes in different plant species varies significantly from one in moss (Physcomitrella patens) to 64 in western poplar (Populus trichocarpa) (Daniel et al., 2015). A. thaliana has 27 TAIR IDs assigned to BBE enzymes, with two splice variants (Supplementary Table 1) (Daniel et al., 2015). Based on the current transcriptome, there are four full length BBE genes (JrBBE1 to 4) that map to different scaffolds in the WGS, in addition to other fragmented transcripts (Table 2 and Table 3). JrBBE1 (C54052_G1_I1) maps to the scaffold JCF7180001213852 and encodes a 564 aa long ORF, which has significant matches to Uniprot:P30986. The closest match of Uniprot:P30986 (with a low significance of 1E-07) to the MAKER-P annotation is ‘WALNUT 00019959-RA’, a 476 aa long cytokinin dehydrogenase. The sequence alignment of JrBBE genes to Uniprot (P30986) is shown (Figure 3a).
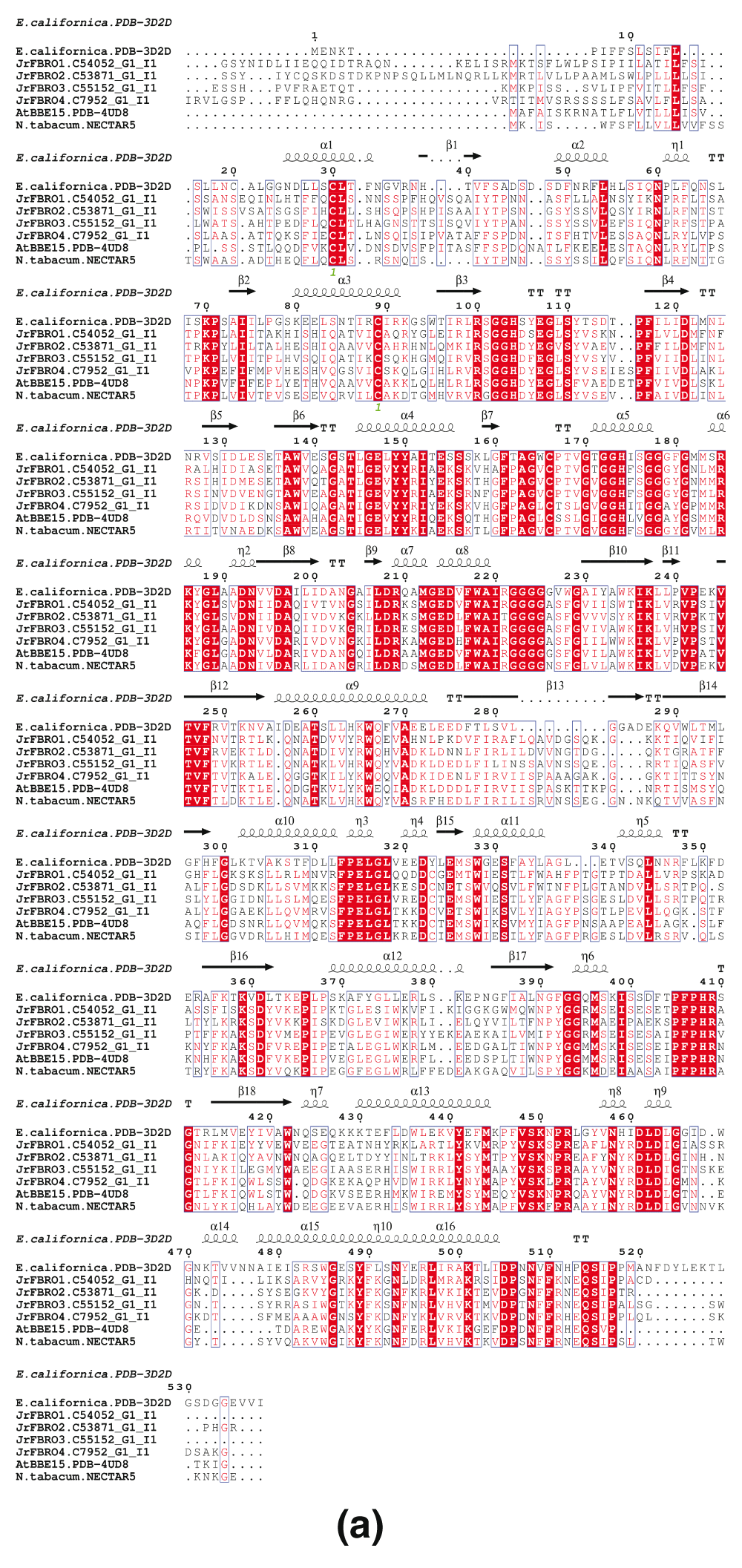

(a) The JrBBE sequences were aligned to berberine bridge enzyme (BBE) genes from Eschscholzia californica (EcBBE; California poppy), Arabidopsis thaliana (AtBBE15) and Nicotiana tabacum (Nectarin V). Secondary structure information from the structure PDBid:3D2D (E. californica) was used to annotate the sequences. The signal peptides are different in these proteins, suggesting different localization of these proteins in walnut. (b) Phylogenetic tree generated from the multiple sequence alignment.
These oxidases are involved in the benzophenanthridine alkaloid biosynthesis in plants. Arabidopsis has 27 loci for this family (and a splice variant) (Table 3). Here, there are four full length berberine bridge enzyme (BBE) genes (named JrBBE1-4) identified using the transcriptome. Some of the proteins are truncated (like C54286_G1_I1), which might be an artifact of the Trinity assembler. Thus, this is not a complete enumeration of the JrBBE genes.
The genes have tissue-specific expression - JrBBE3 is highly expressed in the roots and transition zone. The tissue abbreviations are from Chakraborty et al., 2016a.
As with the walnut transcriptome, the chickpea transcriptome (transHybrid.fasta: n=34760) (Garg et al., 2011) was split into three ORFs, each of which was BLAST’ed to the subset of plant proteins created from the Ensembl database. Subsequently, the ORFs with significant homology to this database (n=29263) were BLAST’ed to the set of annotated chickpea proteins in the NCBI database (n=34198). Most of these annotations were done using Gnomon (Souvorov et al., 2010) (http://www.ncbi.nlm.nih.gov/bioproject/PRJNA190909), which analyzed ~35000 transcripts. There are ~1500 proteins identified by YeATSAM that are absent in the NCBI database (Evalue cutoff 1E-10). Some of these proteins and their corresponding genes in the TAIR database are shown (Table 4). TC00902 is an interesting example with two merged genes: a hydrogen ion-transporting ATP synthase (TAIR ID: ATMG00640.1) and a cytochrome C biogenesis (TAIR ID: ATMG00900.1). While Gnomon identified the cytochrome C biogenesis protein (Genbank: XP_004500083.1), it failed to identify the ATP synthase. Unlike MAKER-P, Gnomon generates transcripts through predictive algorithms and does not take the transcriptome as an input. Notwithstanding, these chickpea genes remain unannotated despite the presence of a straightforward method to detect them from available transcripts.
Most of the NCBI genes were predicted using Gnomon. YeATSAM used the publicly available transcriptome from chickpea to identify these genes. The corresponding genes from the TAIR database are shown. Several transcripts (like TC20962) encode multiple genes, while others (like TC01181) have only one significant ORF. TRid, transcript id; TAIRid: Arabidopsis thaliana id.
Among the ~700 genes not detected by YeATSAM, there are ~500 genes with no matches in the complete ‘nr’ database. Of these, ~300 have no transcripts (SetA), while the remaining ~200 have matches among the transcripts (SetB). Considering the sensitivity of RNA-seq and the wide coverage of twenty tissues, it is a definite possibility that SetA are pseudogenes. Future work in YeATSAM will focus on methods to distinguish these two classes of genes.
The availability of a RNA-seq-derived transcriptome from a newly sequenced organism like walnut, for which there are related annotated genomes (Arabidopsis, Vitis, etc), immensely simplifies annotation of the genome and influences the choice of annotation software. Here, we introduce a new annotation method in the YeATS suite (YeATS Annotation Module - YeATSAM), which was used to annotate the newly-sequenced walnut genome using a simple workstation. The key differentiating factor in YeATSAM is the splitting of the assembled transcriptome into multiple ORFs (Chakraborty et al., 2015). Transcripts often have more than one significant ORF that must be handled differently depending on whether they map to the same or different proteins. We show that YeATSAM failed to annotate ~700 genes identified by MAKER-P, while identifying ~4000 genes missed by MAKER-P. While most of these genes have repetitive stretches, both methods missed vital genes identified by the other. Since many of the additional genes identified by MAKER-P have no known transcripts, we posit that these were identified using ab initio methods. In the absence of such an ab initio module in YeATSAM, we propose a combined method using both MAKER-P and YeATSAM to annotate the WGS. YeATSAM was also applied to the chickpea transcriptome and identified ~1000 proteins that are not annotated in the NCBI database. This transcriptome was assembled using Newbler v2.3 (Garg et al., 2011) and most of the 34198 chickpea proteins in the NCBI database were annotated using Gnomon, the standard annotation tool (http://www.ncbi.nlm.nih.gov/genome/guide/gnomon.shtml).
Latest source code: https://github.com/sanchak/YeATSAM
Archived source code at time of publication: DOI: 10.5281/zenodo.165992 (Sanchak, 2016)
License: GNU General Public License
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)