Keywords
co-expression networks, differential co-expression analysis, biological state transition
co-expression networks, differential co-expression analysis, biological state transition
Recent technological advances have moved the focus of biologists from how to measure biological parameters to how to analyze and interpret tens of thousands of measurements, frequently called omics data. The first solutions for such a problem were limited to hierarchical clustering1–3 and simple comparisons between classes of data through the identification of differentially expressed genes (DEGs)4,5. Nowadays, reconstruction and interrogation of biological networks have become a widely used approach to get insights from different types of omics data6,7.
After establishing co-expression networks for different states of one biological system, differential co-expression analysis investigates their structural changes when a system goes through a state transition. This analysis, first proposed more than a decade ago8,9, identifies the pairs of genes that have their interaction changed during such transition. Several later publications have suggested different algorithms and statistics to determine differential gene co-expression10–27. Fewer studies, however, attempted to evaluate the biological significance of these changes18,21. Also, to the best of our knowledge, there have been no studies that would investigate how this approach performs depending on the type and complexity of the biological system analyzed.
Commonly, a state transition of a biological system is related to perturbation of a set of genes, which propagates through network interactions and affects other genes. Thus, there is a possibility that differentially co-expressed (DC) genes (directly or indirectly) contribute to the propagation of perturbations. In order to investigate the role of DC genes in a state transition of a biological system, we considered two biological processes28,29 previously analyzed by our group. The first one (B cell deficiency in mice) is a homogenous, one-causal-factor process, while the second one (cervical cancer) represents a heterogeneous multi-causal system.
In this work, a co-expression network is an undirected graph, where the set of nodes consists of a set of DEGs, and a pair of nodes is connected if there is a significant correlation between them. Differential co-expression analysis is done by identifying the pairs of genes that suffer significant changes in correlation between two states. Throughout this paper such pairs are called differentially correlated pairs (DCPs) and the genes forming these pairs are considered DC genes.
We started by analyzing the B cell knockout (BcKO) data28, which represents a relatively simple experimental model with only one causal factor (B lymphocytes) and homogenous subject groups since this experiment was performed in highly inbred strains of mice.
In order to select the nodes to reconstruct the co-expression networks (BcKO and Control) we compared gene expression in jejunum between BcKO and control mice and found 509 DEGs (Dataset 1). Next, the edges for each network were determined using significantly correlated pairs of DEGs (Figure 1). To identify DCPs we used the method introduced in21 which compares correlations in the BcKO group and in the Control group. Eighty DCPs were found (Dataset 2), of which 56 represent correlation gains (edges which were not present in Control network but showed up in BcKO) and 24 represent losses.
Now we investigate whether network structural changes, herein represented by DCPs, are related to actual causes of global change in gene expression. In the previous study28, it was shown that intestinal gene expression alterations in BcKO mice are mostly dependent on the ability of B lymphocytes to produce antibodies. Therefore, we analyzed the presence of immunoglobulin coding genes (Ig genes, see Dataset 3) among differentially expressed genes (26 Ig genes among 509 DEGs) in DCPs. We observed that 72% (39 out of 54) of correlation gain DCPs are formed by at least one Ig gene, (Figure 2A). Moreover, we found strong enrichment for Ig genes among DC genes in correlation gain (24% (15 out of 63) of DC genes are Ig genes vs 2.7% (11 out of 415) of other DEGs are Ig genes), while no enrichment was observed for correlation lost as a result of B cell deficiency (Figure 2B). Thus, these results support the idea that differentially expressed genes that acquire correlations during transition from one biological state to another have a high chance to play causal roles in such transition.
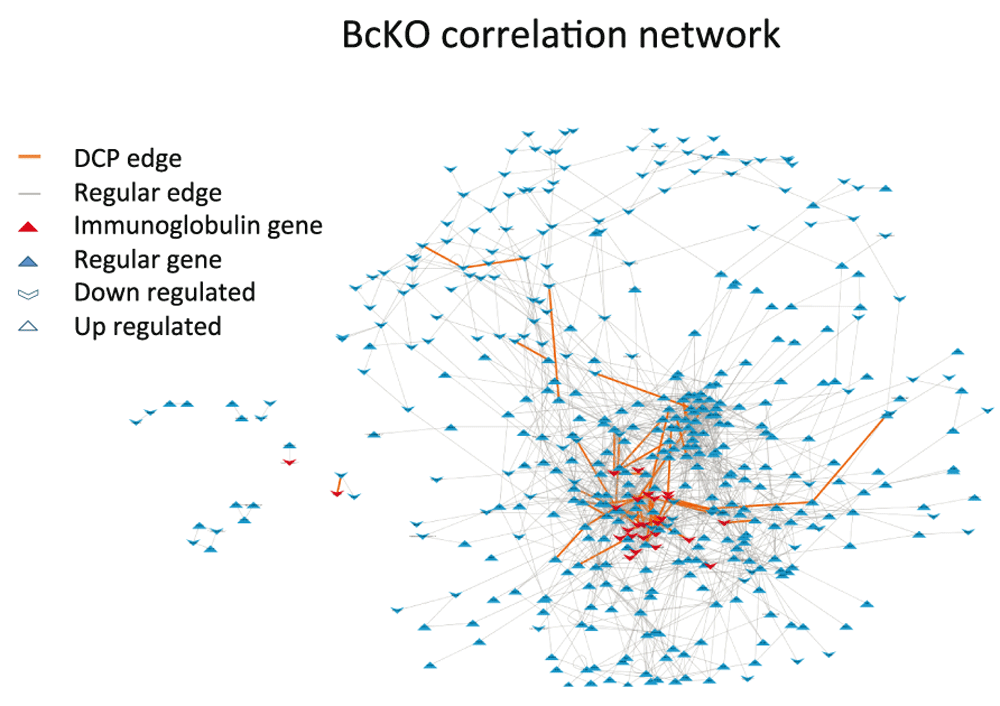
The nodes are composed by DEGs and the edges represent significant correlations between nodes. The causal genes (immunoglobulin genes) and the DCP edges are concentrated in the high connectivity region with several causal genes forming DCPs.

A) 78 Differentially Correlated Pairs (DCPs) were found, of which 54 represent correlation gains (edges which were not present in Control network but showed up in BcKO) and 24 represent correlation losses. The table stratifies the set of pairs representing correlation gains and losses according to the amount of Ig genes (0, 1 or 2) present in a pair. Note that 39 out of 54 of correlation gain DCPs are formed by at least one Ig gene while only 2 out of 22 correlation losses have at least one Ig gene. B) The 78 DCPs are formed by a total of 94 Differentially Co-expressed genes (DC genes). 58 DC genes participate only in correlation gain DCPs, 31 only in correlation loss DCPs and 5 of them participate in both correlation gain and loss DCPs. The results show enrichment for Ig genes among DC genes in correlation gain: 24% (15 out of 63 (=58+5)) of DC genes are Ig genes vs 2.7% (11 out of 415) of other DEGs are Ig genes (p value < 0.001). Meanwhile no enrichment was observed for correlation loss as a result of B cell deficiency: 3% (1 out of 36 (=31+5)) of DC genes are Ig genes vs 2.7% (11 out of 415) of other DEGs are Ig genes.
Analysis of gene expression data. In order to study differentially co-expressed genes in a more complex biological model we turned to cancer. It is well known that cancers of the same clinically/morphological type can be very different on molecular levels. One of the most studied causes for such diversity is the different sets of chromosomal aberrations and mutations harbored by tumors otherwise defined as the same cancer. In previous study29, we have found 36 cervical cancer driver genes located in multiple chromosomal aberrations (Dataset 4). Thus we decided to use cervical cancer data from 29 for investigation of the role of DCPs in complex biological processes due to its heterogeneity and previously acquired knowledge of essential causal genes.
We used the DEGs between tumor and normal tissue as the nodes of the co-expression networks. Since the number of samples (five datasets, 148 tumor samples and 67 normal samples) was larger than in BcKO study (two datasets, 22 paired samples), we used the partial correlation coefficient as a measure of co-expression (Figure 3). The potential advantage of using partial correlation is that it aims to infer edges that are a result of direct regulatory relations6. Partial correlations were calculated through the Local Partial Correlation (LCP) method30 (Material and Methods).
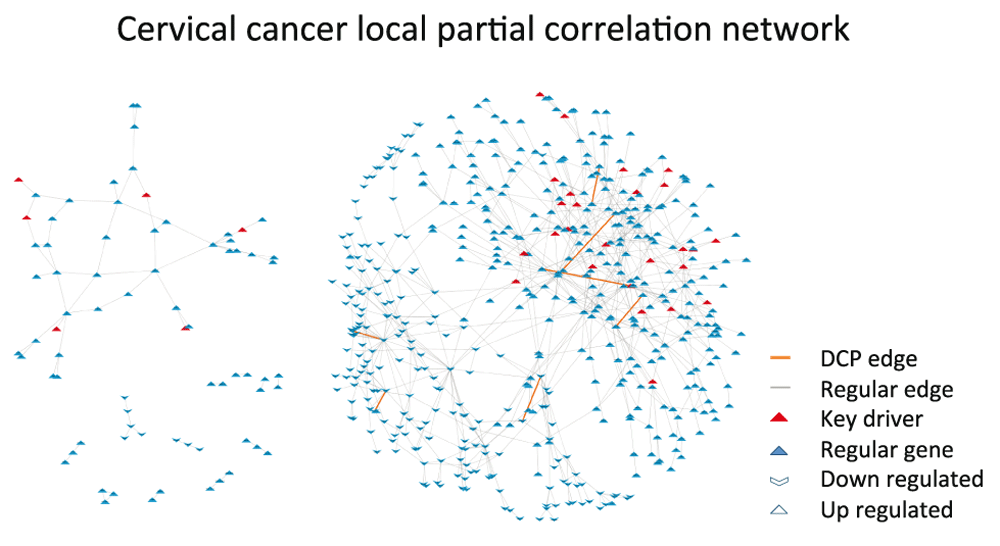
The nodes are composed by DEGs and the edges represent significant local partial correlation between nodes. A few causal genes (key drivers) and DCP edges are located in the high connectivity region, but scattered throughout the network. Only one key driver is amongst the genes in DCPs.
In this study seven DCPs composed of 14 DC genes were found. Interestingly, all DCPs were differential correlations gained in tumors (Table 1). Only one of the 36 key drivers (CEP70) was identified among the 14 DC genes. Accordingly, no enrichment of key driver genes among DC genes was detected in this analysis.
Even though we observed that DCPs are not necessarily formed by key drivers, it is known from literature that most of the DC genes found play regulatory roles in other types of cancer31–48. Thus we hypothesized that DCPs are located downstream of key drivers and can be responsible for changes of regulatory chain events coming from the key drivers and spreading throughout the network. In order to verify this hypothesis, we investigated how close DC genes are to key drivers and whether their “signal flow”49 in the tumor co-expression network is stronger than that of the other genes. In order to verify this hypothesis we investigated two network measures: Minimum Shortest Path and Bi-partite Betweenness Centrality.
First we compared the shortest paths from key driver genes to DC genes and to all other DEGs in the network. We found that DC genes are located statistically closer than the rest of genes in the network to key drivers (Figure 4A, Wilcoxon test < 0.014 and Permutation test < 0.021). Then we used Bi-partite Betweenness Centrality6 as a measure of the signal flow from key drivers to peripheral genes (genes with only one edge)6. We evaluated this measure for DC genes and remaining DEGs and observed that DC genes had much higher values than other genes in the network. Figure 4B illustrates a comparison of boxplots of bi-partite betweenness centrality between these two groups concerning DCPs and the rest (non DCPs, non-key drivers, non-peripheral). We can observe that the bi-partite betweenness centralities of DCPs are concentrated in higher values than the rest. Mann-Whitney test gave us a p-value of 7.868 X 10-5, which gives us evidence that the distribution of Bi-Partite Betweenness Centrality in DCP genes is higher. For more details see Figure S2. Thus, altogether these results suggest that DC genes might be “bottlenecks”, that is, required to transmit a signal from key drivers to other genes in the network, therefore, supplement the hypothesis of a regulatory role of DC genes (Figure S1).
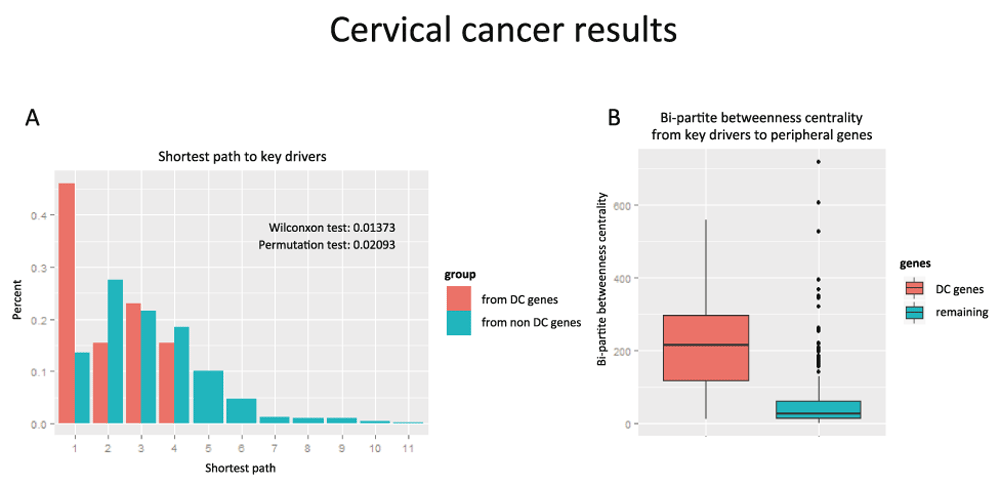
A) Barplot of the shortest path to the causal genes and originated in either the genes in DCPs (in orange) or the non DCP genes (in blue). The distribution in orange is concentrated in lower values. B) Boxplot comparing the values of Bipartite Betweenness Centrality of the genes in DCPs (in orange) and the non-DCP genes (in blue). The boxplot on the left is concentrated in higher values.
Knockdown experiments. In addition, data from other cancers provide indirect support for the idea of regulatory role of DC genes in cervical cancer31–48. However, since a role of these DC genes in carcinogenesis was not as straightforward as for immunoglobulin genes in B cell deficiency, we decided to perform experimental tests. Among the DC genes found for cervical cancer, there were seven up-regulated and seven down-regulated in cancer. Therefore, for validation experiments we chose one down-regulated (FGFR2) and one up-regulated (CACYBP) gene that have not been previously studied in cervical cancer for regulatory properties, but have a potential connection with cell death or proliferation based on their Gene Ontology annotations. In order to test if FGFR2 and CACYBP play critical regulatory roles in cancer pathogenesis, we evaluated the effect on in vitro knockdown of these genes on cell proliferation in a cervical carcinoma cell line.
First, we tested two cervical cancer cell lines (Hela and ME180) and found that only ME180 had detectable expression levels of both genes. In order to perform these tests, we evaluated siRNAs and observed that they were able to knock down expression of both genes by at least 70% (Figure 5A). CACYBP is up-regulated in tumor tissue, as compared to normal tissue (Figure 5B). Consequently, if CACYBP has regulatory potential, as predicted by our analysis, it should function as an oncogene promoting cell proliferation. Therefore, the knockdown of this gene should result in a decrease of cell growth/survival. Since FGFR2 was found down-regulated in cervical carcinomas (Figure 5B) its potential regulatory role would be as a tumor suppressor. Therefore, the knockdown of this gene is expected to increase cell growth. The subsequent analysis of cell proliferation confirmed our predictions for both genes: knockdown of CACYBP led to a decrease of cell growth, while knockdown of FGFR2 induced higher cell proliferation (Figure 5C). Thus, these results provide additional support to our in silico prediction that DC genes may play a regulatory role in cell proliferation related to tumor growth.

A) Efficacy of FGFR2 and CACYBP siRNA knockdown. qRT-PCR with primers for GAPDH as the internal control was used to determine expression and efficacy of FGFR2 and CACYBP specific siRNA knockdown in endothelial cells (ME180). ME180 cells were harvested 72 h after transfection with vehicle (Lipofectamine) and either scrambled control or targeting siRNA. B) Gene expression of FGFR2 and CACYBP (mean +/- standard deviation) for tumor and normal samples from five datasets used in the analysis. Since FGFR2 was found down-regulated in tumor tissue, its potential regulatory role would be as a tumor suppressor. However, CACYBP is up-regulated, thus CACYBP should function as an oncogene promoting cell proliferation. C) Evaluation of cell proliferation inhibition using xCelligence System. Proliferation data (cell index) was obtained at 72 h after transfection with Lipofectamine and either scrambled control or targeting siRNA. Inhibition index was calculated (two step normalization of cell index): inhibition index > 0 – cells transfected with targeting siRNA showed decrease in proliferation; < 0 – showed increase in proliferation; = 0 – no difference from control was found. One sided T test for mean (< 0 for FGFR2 and > 0 for CACYBP) was applied and returned statistically significant p-values for both of them (0.0258 for FGFR2 and 0.01978 for CACYBP).
In the current study, the differential co-expression analysis21 was applied to two relatively well-investigated biological systems28,29 in order to evaluate the potential importance of genes found using differential correlation analyses. Overall, the obtained results support the idea that DC genes play a regulatory role. While in B cell deficiency DCPs were found highly enriched with immunoglobulin genes (i.e. causal genes for alterations in the gut) we did not observe enrichment for key driver genes in cervical cancers. Rather, DCPs of cervical cancer seem to be located downstream of causal genes. Indeed, those DCPs have been found closer to key regulators than other genes in the network, actually representing “bottlenecks” for communication between driver genes previously published in 29 and the rest of the network (Figure 4). Furthermore, some differentially co-expressed genes in cervical cancer have been previously implicated in processes such as metastasis, oncogenic autophagy and apoptosis. For example, CACYBP has been shown to promote colorectal cancer metastasis31, TRPM3 was observed to play a role in oncogenic autophagy in clear cell renal cell carcinoma32,33, and AK2 was reported to activate apoptotic pathway34. Several genes are investigated for prognostic value for cancers such as myeloma35, lymphoma36, breast37–41 and gastrointestinal42,43 cancers. At least two genes were previously proposed as targets for anti-cancer agents: DHFR44 and FGFR245. Moreover, CACYBP and ZSCAN18 were also reported as putative tumor suppressor genes in renal cell carcinoma30,46,47. In addition, we have tested two DC genes and confirmed their regulatory role (FGFR2 as a tumor suppressor and CACYBP as a potential oncogene in cervical cancer) by manipulating their expression in vitro. Altogether, published observations and our experimental validation for these two genes support the idea that DC genes revealed in the current study play a regulatory role and can be candidate targets for cervical cancer treatment.
Interestingly, while in the model of B cell deficiency, the DC genes are highly enriched with causal regulatory genes, there was only one key driver in cervical cancer (CEP70), despite the DC genes in this system still seeming to play a regulatory role overall. Such a difference is potentially related to the fact that the mouse system studied in 28 is highly homogeneous (inbred mice) with only one cause of alterations (i.e. absence of B lymphocytes). Cervical cancer, however, is a heterogeneous system with different chromosomal aberrations and consequently turned-on expression of different driver genes in different patients. Therefore, we can speculate that differential correlations point to regulatory genes that are shared by majority of samples. This hypothesis warrants further investigation, especially considering that DCPs could represent common therapeutic targets for tumors that originated as a result of different genomic or epi-genomic events.
In conclusion, this study provided additional evidence for the previously suggested idea8–27 that genes presenting alterations in correlation patterns between different phenotypes (i.e. states of biological system) play a critical regulatory role in transitions from one state to another. Furthermore, although our results do not allow for full generalization, they indicate that gain and not loss of correlations connects critical genes involved in transitions to new phenotypes. However, further studies are required to understand how changes in correlation patterns can point to genes with critical capacity to guide a biological system into certain state/phenotype.
BcKO. All microarray data were analyzed using BRB Array-Tools developed by the Biometric Research Branch of the National Cancer Institute under the direction of R. Simon (http://linus.nci.nih.gov/BRB-ArrayTools.html). Array data were filtered to limit analysis to probes with greater than 50% of samples showing spot intensities of >10 and spot sizes >10 pixels, and a median normalization was applied.
Cervical cancer. Same as in cervical cancer29. The data were analyzed using BRB Array-Tools using the original normalization used in three studies50–52 and median normalization over entire the array for the fourth study53. For all studies, we only considered genes found in at least 70% of arrays.
In every analysis (DEGs, DCPs and networks), filter of direction (same sign of correspondent parameter – difference of mean, difference of correlation, correlation and partial correlation) was required in a fixed number of datasets (2 out of 2 in BcKO and 3 out of 5 in cervical cancer). Then meta-analysis was done through Fisher combined probability test54. Next, the pairs with false discovery rate (fdr)55 lower than a threshold are chosen. At last, only the pairs that pass PUC56 are considered correlated and therefore represent edges in the network.
BcKO. DEGs between groups of samples were identified by random variance paired t-test p-value lower than 5% with adjustment for multiple hypotheses by setting the fdr below 10% in BRB Array-Tools. Co-expression networks (BcKO and Control) were inferred through Pearson correlation with p-value < 20% and fdr adjustment below 2.5%. DCPs were calculated for pairs that were initially correlated (p-value < 20%) in at least one state. Then differences of Pearson correlation were tested following21 with a p-value below 10% and fdr < 2%. At last only the DCPs that showed up in one of the networks were selected.
Cervical cancer. DEGs were retrieved from a cervical cancer paper29. Correlation networks and DCPs followed the same procedure and in BcKO but with different p-values (correlation p-value < 10% with fdr < 10-8 and difference of correlation p-value < 10% with fdr < 0.25%). Partial correlation was computed using local partial correlation method30. The initial significance was p-value lower than 40% and then fdr < 5%.
For more details about the thresholds used, see Table S3 and Table S4.
Two aspects of cervical cancer data motivated us to use local partial correlation for this system. First of all, we have more samples throughout five datasets (see Supplementary Table S1 and Supplementary Table S2) which allows us to have more confidence in our results and second we already know that tumors in general present heterogeneous causal factors. The partial correlation approach gives us the alternative to only consider edges that represent direct regulatory relations.
In this paper we used the new approach developed in 30 called local partial correlation. This approach was elaborated specially for cases when there are more variables than samples, which happens regularly in genetics and is a serious problem in classical statistics. First we calculate the correlation network. Then for each significantly correlated pair the inverse method is applied exclusively to the correlation sub-matrix formed only by the closest neighbors of the pair along with the genes forming the pair, Figure 6. If the number of closest neighbors is still higher than the number of samples n, then we decreasingly rank the correlations of the neighbors to either genes in the pair and select the first n/2 neighbors. For each sub- matrix, we only keep the partial correlation value regarding the pair that formed that sub- matrix and then calculate its p-value also based on the sub- matrix. R script for calculation is available in Supplementary Material.
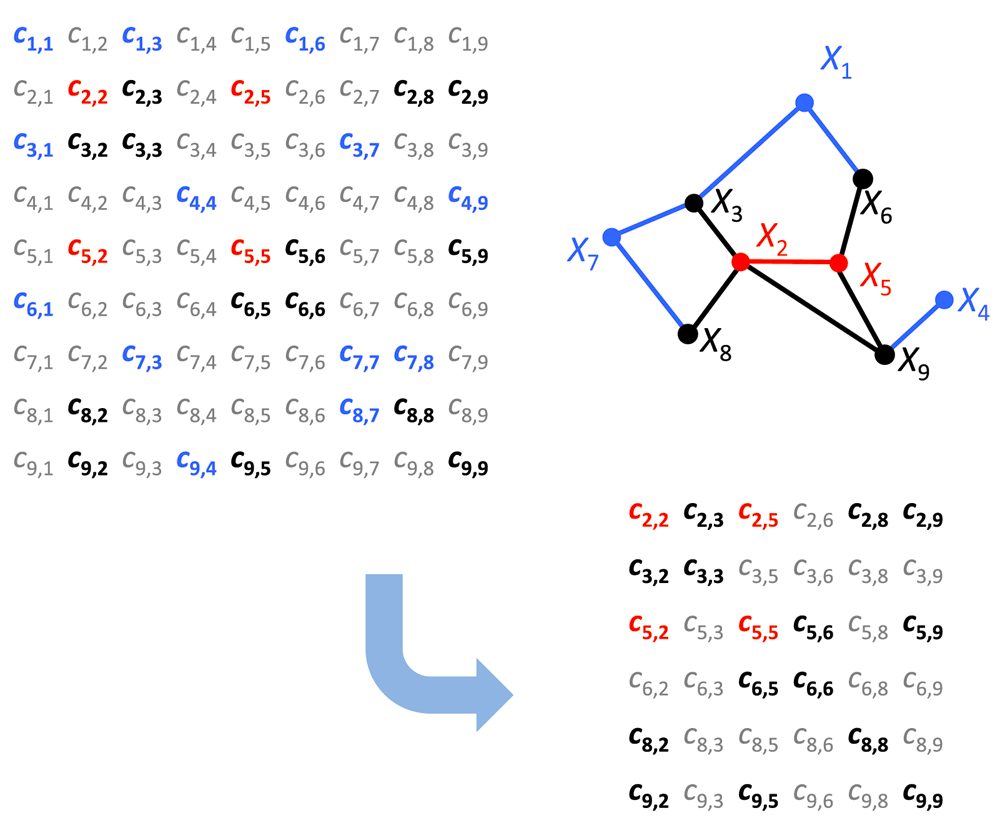
The neighborhood of this pair is the set of nodes X3, X6, X8, X9 (black nodes/edges). X1, X4, X7 (blue nodes) are significantly correlated with the black nodes (blue edges), but not with the red nodes. Thus the inverse method is applied exclusively to the correlation sub-matrix formed only by the genes X2, X5, X3, X6, X8, X9. In correlation matrices the gray entries are statistically non-significant empirical correlations.
Partial correlations were estimated only for the significant (Pearson) correlations in co-expression network. Thus the same definition of DCPs (by Pearson correlation) can still represent structural changes as long as it remains present in one of the two networks.
Figure 3 illustrates the local partial correlation network for cervical cancer using only tumor data. It has 578 connected nodes and 824 edges.
The shortest path is a method that calculates distances between 2 nodes in a network. It consists of the minimum number of edges connecting 2 nodes. In this case we want to know the minimum number of edges connecting one node, either DCP gene or not, to a group of nodes: the key drivers Figure 7. For each gene we calculate the shortest path to all key drivers and get the minimum value. Then we compare the minimum shortest path to key drivers coming from DCP genes and the remaining genes. Figure 4A shows that the minimum shortest path to key drivers tend to be smaller when originated in DCP genes.
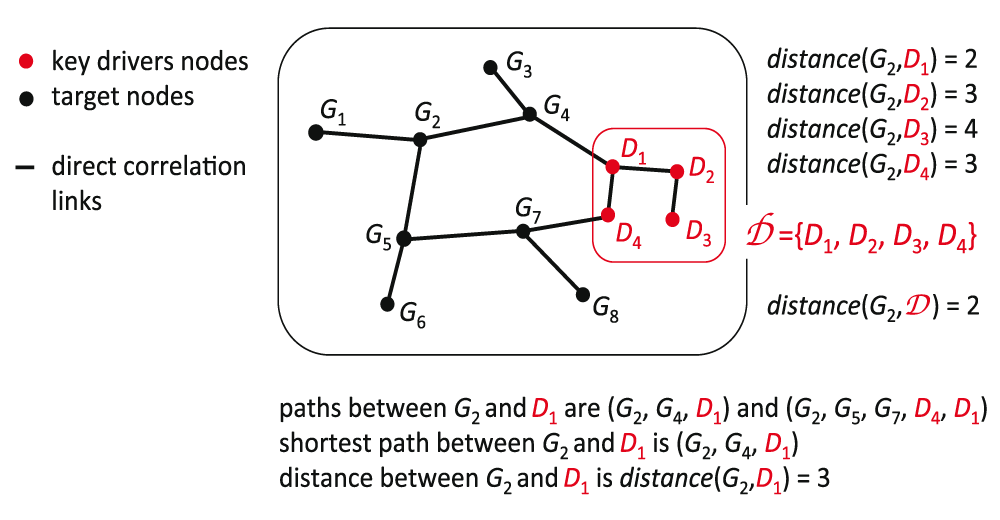
Betweenness Centrality measures the node’s centrality in a network by counting the number of shortest paths from all vertices to all other vertices that pass through that node. A gene with high betweenness centrality has a great influence on the transfer of signal through the network Figure 8.
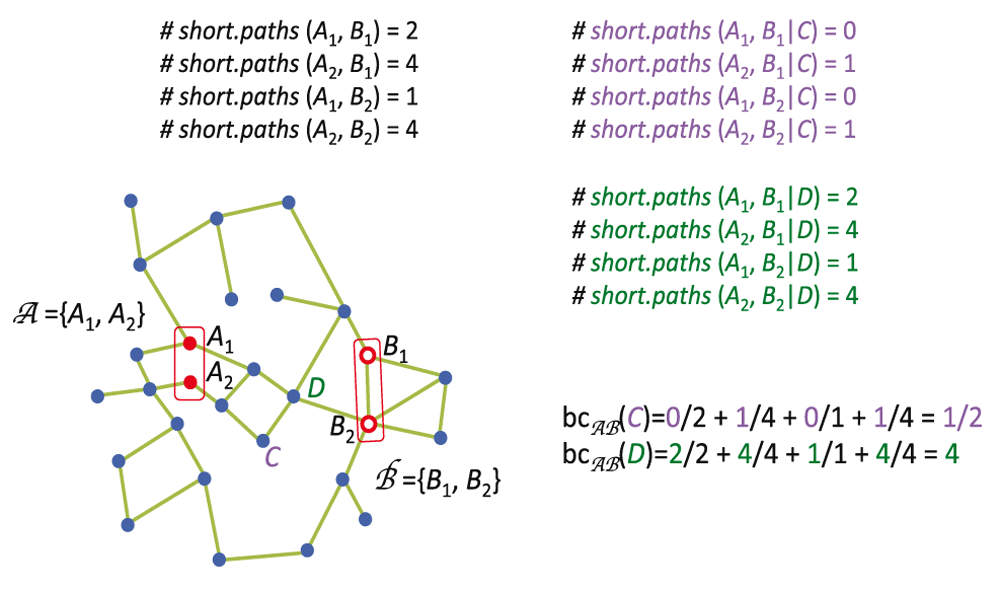
Note that the node D has bigger bc because all shortest paths connecting nodes in group A to nodes in group B pass through the node D.
However we are interested in the signal passing from key drivers throughout the network. For this reason we decided to apply the measure previously developed by our lab6 called Bi-partite Betweenness Centrality. It measures the amount of shortest path going from all genes in one group of vertices to all genes in a different group of vertices. In our case, the groups of genes are the key drivers and the peripheral genes (genes connected to only one edge).
ME180 cells were transfected with FGFR2-, CACYBP-specific siRNA or control siRNA using Lipofectamine RNAiMAX Transfection Reagent. Cell growth rate during 72h after siRNA transfection was measured using xCelligence system as described below.
Evaluation of siRNA efficacy in knocking down the gene targets. ME180 cell line was obtained from Dr. Pulivarthi H. Rao. It was cultured in RPMI medium with 10% FBS and 1% Penicillin-Streptomycin added. The cells were seeded at density 4000 cells per well in 96 F-bottom plates (seeding procedure was done according to ATCC protocol for ME180 cell line) and with cell culture media 200 ul per well. 24 hours after seeding, cells were transfected with one of the three siRNA, see Table 2.
| Target | Supplier | Supplier ID |
|---|---|---|
| FGFR2 | ThermoFisher | s5173 |
| CACYBP | ThermoFisher | s25819 |
| Non-targeting siRNA | Dharmacon | D-001810-01-05 |
Before transfection, 100 uL of media was taken from each well. Transfection procedure was done according to Lipofectamine RNAiMAX Reagent protocol (Protocol Pub. No. MAN0007825 Rev. 1.0). 3pM of siRNA per well and Lipofectamine 0.6 uL per well were delivered in 20uL. 80 uL of fresh cell culture media was added to each well.
Cells were collected 72 h after transfection using Lysis buffer from RNeasy Mini Kit (QIAGEN). RNA extraction was done using RNeasy Mini Kit (QIAGEN) according to the manufacturer’s protocol (no Dnase treatment step was done). Concentrations of RNA measured with Qubit RNA BR Assay Kit. cDNA was done using Bio-Rad iScript cDNA Kit according to the manufacturer’s protocol.
Quantitative Real-Time PCR was done for the samples using QuantiFast SYBR Green PCR Kit and GAPDH as a control gene. Primers for the targets you can see in the Table 3.
qRT PCR set up: sample was heated to 95°C, followed by 40 cycles of 95°C for 10 sec and 60°C for 30 sec.
Evaluation of cell growth after knock down of gene targets. CACYBP is up-regulated in tumor tissue, as compared to normal tissue (Figure 5B). Consequently, if CACYBP has regulatory potential, as predicted by our analysis, it should function as an oncogene promoting cell proliferation. Therefore, the knockdown of this gene should result in a decrease of cell growth/survival. Since FGFR2 was found down-regulated in cervical carcinomas (Figure 5B) its potential regulatory role would be as a tumor suppressor. Therefore, the knockdown of this gene is expected to increase cell growth.
Cell growth was evaluated using xCelligence system (The RTCA DP Instrument) using manufacturer’s protocol. ME180 was cultured in RPMI media with 10% FBS and 1% Penicillin-Streptomycin added. The cells were seeded at density 4000 cells per well (E-Plate 16) in 200 uL of cell culture media.
24 hours after seeding, the experiment was paused for transfecton. Before transfection, 100 uL of media was taken from each well. Transfection procedure was done according to Lipofectamine RNAiMAX Reagent protocol (Protocol Pub. No. MAN0007825 Rev. 1.0). 3pM of siRNA per well and Lipofectamine 0.6 uL per well were delivered in 20uL; 80 uL of fresh cell culture media was added to each well. Plate was placed back in the slot and cell growth was evaluated for another 72 h.
Cell index normalization. To evaluate cell growth rate cell index was transformed into Inhibition index in two steps:
1. Cell indexes for all wells were exported to the excel file. For each treatment (including non-targeting siRNA transfected wells) we extracted cell index average for all wells at 20 h after seeding (Cell Index Before Treatment) and at 96 h after seeding (Cell Index After Treatment). To normalize cell index to initial cell number differences for each of the treatments we used the following formula:
2. In next step we normalized each treatment with targeting siRNA to treatment with non-targeting siRNA. For this purpose in each experiment A/B Index from treatment (siRNA targeting either FGFR2 or CACYBP) was normalized to A/B Index from control treatment using the following formula:
Final evaluation of growth was done according to the value of Inhibition Index:
>0 – there is a decrease in growth;
0 – no difference between treated with targeting and treated with non-targeting siRNA;
<0 – there is a growth after treating with targeting siRNA.
BcKO: Gene expression files containing array data from 28 are available under the GSE23934 superseries in the Gene Expression Omnibus (GEO) data repository. We worked with two groups of samples: B10.A littermates and BALB/C (Table S1).
Cervical cancer: We have used the same datasets as in previous study29 available at GEO: GSE741050, GSE679151, GSE780352, GSE975053, GSE2634229 (Table S21).
F1000Research: Dataset 1. Differentially expressed genes from BcKO study, 10.5256/f1000research.9708.d14210057
F1000Research: Dataset 2. Differentially correlated pairs from BcKO study, 10.5256/f1000research.9708.d14209958
F1000Research: Dataset 3. Causal genes from BcKO study, 10.5256/f1000research.9708.d14209759
F1000Research: Dataset 4. Causal genes from cervical cancer study, 10.5256/f1000research.9708.d14209860
F1000Research: Dataset 5. Cytoscape Edges and Nodes tables from network in Figure 1, 10.5256/f1000research.9708.d14210161
F1000Research: Dataset 6. Cytoscape Edges and Nodes tables from network in Figure 3, 10.5256/f1000research.9708.d14210262
F1000Research: Dataset 7. Raw data for Figure 5A,C, 10.5256/f1000research.9708.d14210363
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)