Keywords
Plasmodium falciparum, Plasmodium vivax, non-synonymous polymorphism, resistance evolution
Plasmodium falciparum, Plasmodium vivax, non-synonymous polymorphism, resistance evolution
Plasmodium falciparum and P. vivax are the two major species causing malaria in humans. These species differ greatly in their geographical distribution, mortality rates and resistance to anti-malarial drugs. P. falciparum is responsible for ~200 million malaria cases and ~440,000 deaths annually, of which ~90% occur in Africa, while P. vivax causes ~14 million malaria cases and 1400–15000 deaths annually, of which ~75% occur in South and South-East Asia1. Chloroquine was used as a frontline drug for both P. falciparum and P. vivax, but widespread resistance to chloroquine has only been observed in P. falciparum2. Therefore, chloroquine remains a frontline drug against P. vivax in most parts of the world, despite its usage for ~70 years2. The crt gene, which is involved in chloroquine resistance in P. falciparum, is not associated with chloroquine resistance in P. vivax3, despite significant conservation of the protein in the two species, and the mechanism of chloroquine resistance in P. vivax remains unknown. P. falciparum strains have developed resistance to almost all currently used drugs, including artemisinin, the most effective anti-malarial drug, and dealing with drug-resistant P. falciparum is one of the main contemporary public health challenges4,5. While P. vivax has been exposed to artemisinin, due to its frequent co-infection with P. falciparum6 and the usage of artemisinin against P. vivax in areas with chloroquine resistance7, artemisinin resistance in P. vivax has not yet been observed8–10. Thus, the evolutionary response of P. falciparum and P. vivax against anti-malarial drugs appears to be different11.
A large amount of genome sequencing data has recently been generated from thousands of P. falciparum and hundreds of P. vivax samples12–14. This provides an unprecedented opportunity to compare the evolutionary patterns in the two species. The present study analysed this genomic data, and found a large substitution bias in P. vivax, even at non-synonymous sites, leading to biased amino acid changes. This may be related to the differential evolutionary response to same anti-malarial drugs observed in the two parasites.
The single nucleotide polymorphism (SNP) data of P. falciparum and P. vivax was obtained from the MalariaGen community webpage (https://www.malariagen.net/data/p-falciparum-community-project-jan-2016-data-release; https://www.malariagen.net/data/p-vivax-genome-variation-may-2016-data-release). The SNP data for P. falciparum consists of filtered and high quality 939,687 exonic SNPs with 631,715 non-synonymous and 307,972 synonymous SNPs from 3,394 samples from 22 countries13. The SNP data for P. vivax consists of filtered and high quality 303,616 SNPs from 228 samples14. Of these there were 87,877 non-synonymous, 62,862 synonymous and 152,877 non-coding SNPs. Proteome sequences of P. falciparum 3D7, P. berghei ANKA, P. chabaudi chabaudi, P. cynomolgi B, P. knowlesi H, P. reichenowi CDC, P. vivax Sal1, P. yoelii 17X were downloaded from the PlasmoDB database (http://plasmodb.org/common/downloads/release-27/). Orthologous sequences were identified using best bidirectional hit algorithm15 and aligned using ClustalO (http://www.clustal.org/omega/)16. The conservation score for P. vivax residues was calculated as the average substitution score using BLOSUM62 matrix across seven orthologs at non-gapped positions.
All statistical analyses were performed in R software v3.3.1 (https://www.r-project.org/). R commands cor.test was used for calculating the Spearman rank correlation coefficients.
There is a large difference in the genomic AT content of the two Plasmodium species. P. falciparum has a genomic AT content of 81% compared to 58% for P. vivax, thus the two species have diverged in their AT content from their common ancestor17. It has been proposed that the common ancestor of the two species was AT rich17 and P. vivax has increased its genomic GC content since its divergence from the common ancestor. We tested whether this is true during the recent evolution of P. vivax by analysing the SNP data. We found highly biased substitution patterns in P. vivax, such that SNPs that change GC to AT nucleotides were approximately three times more common than those that change AT to GC nucleotides (Figure 1). This bias was present at synonymous, non-synonymous and non-coding sites (Figure 1) and indicates a recent opposite substitution bias in P. vivax compared to the general increase in its genomic GC content since its divergence from the common ancestor of P. falciparum and P. vivax. The biased substitution pattern at non-synonymous sites in P. vivax was reflected in the pattern of amino acid changes at the polymorphic sites, such that amino acids with GC-rich codons are reduced in abundance, while amino acids with AT-rich codons are increased in abundance (Figure 2A).
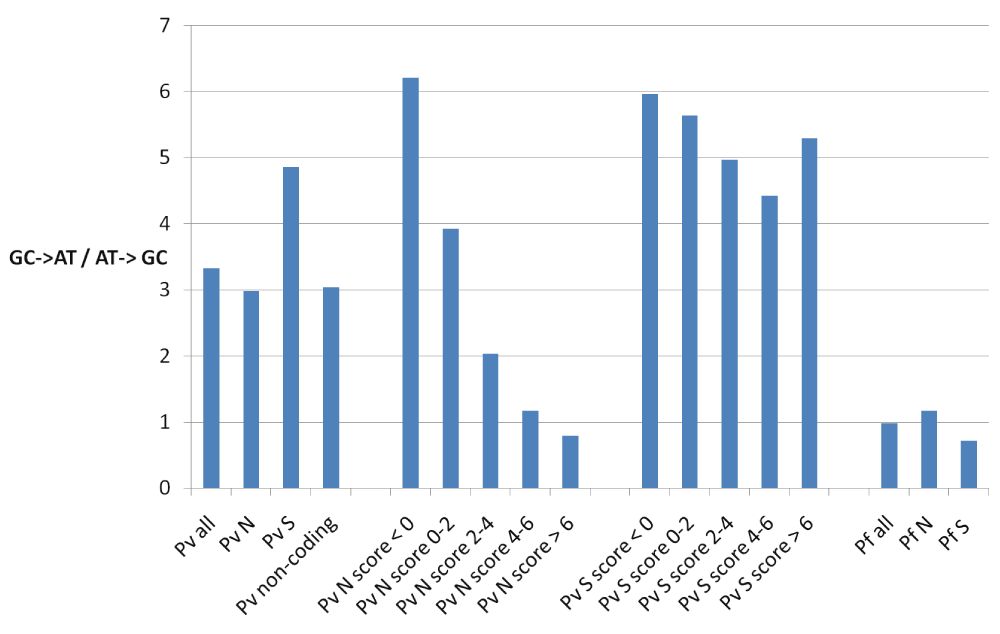
There are three times as many single nucleotide polymorphisms in P. vivax (Pv) that change GC to AT nucleotides compared to those that change AT to GC nucleotides. This bias becomes higher at synonymous sites (S). At non-synonymous sites (N) the bias becomes lower with a higher conservation score. The conservation score for each amino acid at non-synonymous sites was calculated as the average BLOSUM62 substitution score across seven Plasmodium orthologs at non-gapped positions. No such bias was observed in P. falciparum (Pf).
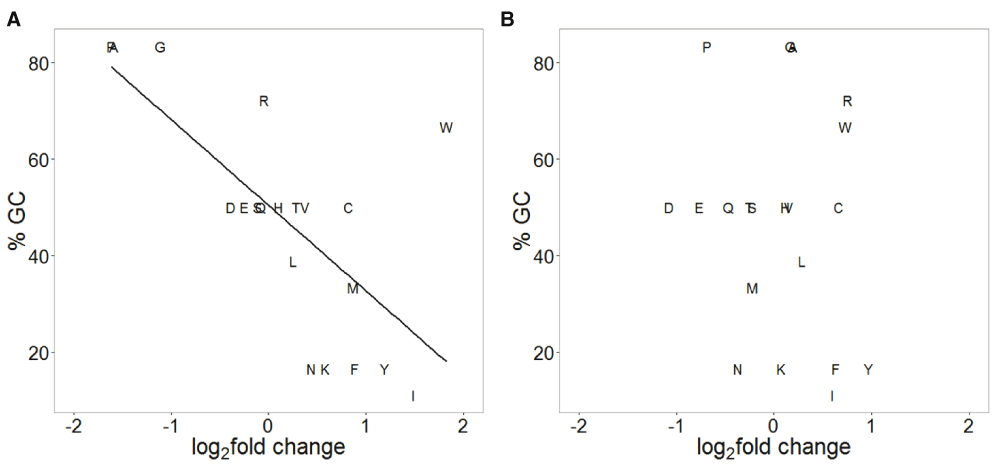
(A) P. vivax. The average % GC content of codons for different amino acids is plotted on the Y-axis and the log2 fold change in the amino acid abundance at non-synonymous sites is plotted on the X-axis. A significant negative correlation is observed (Spearman correlation coefficient 0.69, p=0.0008). (B) P. falciparum. No correlation was observed (Spearman correlation coefficient -0.09, p=0.7).
We asked whether substitution bias in P. vivax might also influence amino acid changes at conserved positions. The substitution bias was present at amino acid positions that are low to moderately conserved, but not at highly conserved positions (Figure 1). As a control, there was no relationship between conservation and substitution bias at synonymous sites (Figure 1).
We next tested whether similar bias might be present in P. falciparum. There was no marked substitution bias at synonymous or non-synonymous sites in P. falciparum (Figure 1). Consequently, there was no bias in amino acid changes at polymorphic sites according to the GC content of its codons (Figure 2B).
The present study finds a sharp recent reversal in the substitution bias in P. vivax favouring AT nucleotides compared to the general increase in its GC content since its divergence from the common ancestor of P. vivax and P. falciparum. This substitution bias has a consequence for the pattern of amino acid changes even at moderately conserved, and thus functionally important, sites (Figure 1). No such bias was observed in P. falciparum (Figure 1). The large difference in the substitution bias between P. vivax and P. falciparum may lead to different evolutionary solutions to similar drug pressure. It has been proposed that differences in the life cycle of the two Plasmodium species, specifically the early onset of gametocyte stage in P. vivax, which allows transmission before the malaria symptoms and drug treatment, may impede the spread of drug resistance in P. vivax11. It is also possible that the strength of negative selection might be different between the two species. The ratio of non-synonymous to synonymous polymorphisms (N/S) is much higher in P. falciparum compared to P. vivax (2.113 and 1.414, respectively). We found that the difference in N/S was also present when considering amino acid sites conserved across Plasmodium species (0.64 and 0.37 for P. falciparum and P. vivax, respectively). A higher tolerance for non-synonymous changes at conserved amino acid positions in P. falciparum suggests that fitness reducing mutations might have a higher likelihood to be established in P. falciparum compared to P. vivax. Since drug resistance evolution often entails fitness cost18, it might be easier to acquire fitness reducing drug resistance mutations in P. falciparum compared to P. vivax. It is likely that a combination of these and other factors might contribute towards differences in the drug resistance evolution in the two species.
It has been proposed that the common ancestor of P. falciparum and P. vivax was AT-rich and P. vivax has subsequently been evolving towards higher GC content17. Here we find a recent reversal in the substitution bias in P. vivax, where it is now evolving towards increasing AT content. Interestingly, we observed a lower substitution bias in the non-coding regions in P. vivax compared to synonymous sites (Figure 1), which might suggest a higher functional constraint in the non-coding regions compared to synonymous sites. This observation may be utilized to identify non-coding regions in P. vivax genomes that are under higher functional constraint as more genomics data becomes available in the future.
This publication uses data from the MalariaGEN Plasmodium falciparum Community Project, as described in ‘Genomic epidemiology of artemisinin resistant malaria’, eLife, 2016 (DOI: 10.7554/eLife.08714)13, and the MalariaGEN P. vivax Genome Variation project, as described by Pearson et al. in Nature Genetics, 2016 (DOI: 10.1038/ng.3599)14. This data is also available from the MalariaGEN website (https://www.malariagen.net/data/p-falciparum-community-project-jan-2016-data-release; https://www.malariagen.net/data/p-vivax-genome-variation-may-2016-data-release).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)