Introduction
The recent regulatory approval of venetoclax for the treatment of chronic lymphocytic leukemia (CLL) culminates 30 years of investigation in many labs worldwide. Milestones in this effort have included the cloning of BCL2 at the t(14;18) translocation in follicular lymphomas1,2, demonstration that BCL2 inhibits cell death3,4, realization that BCL2 is elevated in CLL5,6, recognition that BCL2 and its anti-apoptotic paralogs bind BH3-only proteins through their BH3-binding grooves7, identification of ABT-737 and navitoclax as BH3-binding groove-directed inhibitors of BCL2 and BCLXL8,9, demonstration that navitoclax is active against CLL10, and derivation of venetoclax as a BCL2-selective BH3 mimetic11. While the approval of venetoclax for CLL is a triumph in its own right, the challenge remains to optimize the use of this agent and other BH3 mimetics for improved therapy of diverse malignancies. To provide context for these ongoing efforts, we review recent progress in understanding the action of BCL2 family proteins, summarize the clinical status of venetoclax and other BH3 mimetics, and discuss possible approaches to predicting whether various cancers will respond to these agents.
Mitochondrial apoptosis and BAX/BAK activation
BH3 mimetics are designed to inhibit anti-apoptotic BCL2 family proteins, leading to BAX and BAK activation12–14. Accordingly, recent advances in understanding the functions of various BCL2 family members provide important insight into the therapeutic effects of BH3 mimetics.
Mitochondrial apoptosis
BCL2 family members regulate apoptosis, a distinct form of cell death that plays critical roles in development, immune response, and tissue homeostasis15–17. This type of cell death can be triggered through two different pathways depending on the stimulus. The death receptor pathway is initiated through binding of death ligands to certain cell surface receptors. In contrast, the mitochondrial or intrinsic apoptotic pathway involves the release of mitochondrial intermembrane proteins, including cytochrome c and Smac/Diablo, to the cytosol, where they contribute to subsequent apoptotic changes18–20. The translocation of these intermembrane proteins is modulated by the BCL2 family of proteins.
Based on differences in structure and function, BCL2 family members are divided into three subgroups20–22: BAX and BAK, which contain three distinct BCL2 homology (BH) domains and, upon activation, permeabilize the mitochondrial outer membrane (MOM) by forming proteinaceous pores23–26 or in other ways27–30; the anti-apoptotic family members BCL2, BCLXL, MCL1, BCLW, and BCL2A1 (also called BFL1 in humans and A1 in mice), which typically contain four BH domains and oppose MOM permeabilization; and the BH3-only proteins BIM, BID, PUMA, NOXA, BAD, BIK, BMF, and HRK, which share homology with other BCL2 family members only in their 15-amino-acid α-helical BH3 domain and induce apoptosis by facilitating BAX and/or BAK activation22.
BAX/BAK activation models
Three different models have been proposed to explain BAX and BAK activation. The direct activation model proposes that certain BH3-only proteins directly interact with BAX and/or BAK to cause a conformational change that leads to BAX/BAK oligomerization and activation31–33. In this model, the major role of anti-apoptotic BCL2 family members is to inhibit the BH3-only proteins. The indirect activation model proposes that BAX and BAK are tonically activated but are restrained by anti-apoptotic BCL2 family members34. In this model, BH3-only proteins induced by various death signals primarily inhibit the anti-apoptotic BCL2 family members, leading to the release of activated BAX and BAK. Finally, the unified model proposes that anti-apoptotic BCL2 family proteins inhibit both BH3-only proteins and activated BAX or BAK35. In both instances, the exposed BH3 domains of the pro-apoptotic proteins are neutralized by interaction with BH3-binding grooves, extended clefts on the surfaces of anti-apoptotic BCL2 family members36,37. The BH3 mimetics described below have been identified and developed based on their ability to occupy the same BH3-binding grooves.
Two mechanisms of BH3 mimetic-induced killing
Neutralization of BH3-binding grooves on anti-apoptotic BCL2 family members is not, by itself, sufficient to kill cells. Instead, binding of BH3 mimetics to anti-apoptotic BCL2 family members must result in BAX and/or BAK activation to elicit cell death. This BAX/BAK activation can occur by one of two processes (Figure 1).
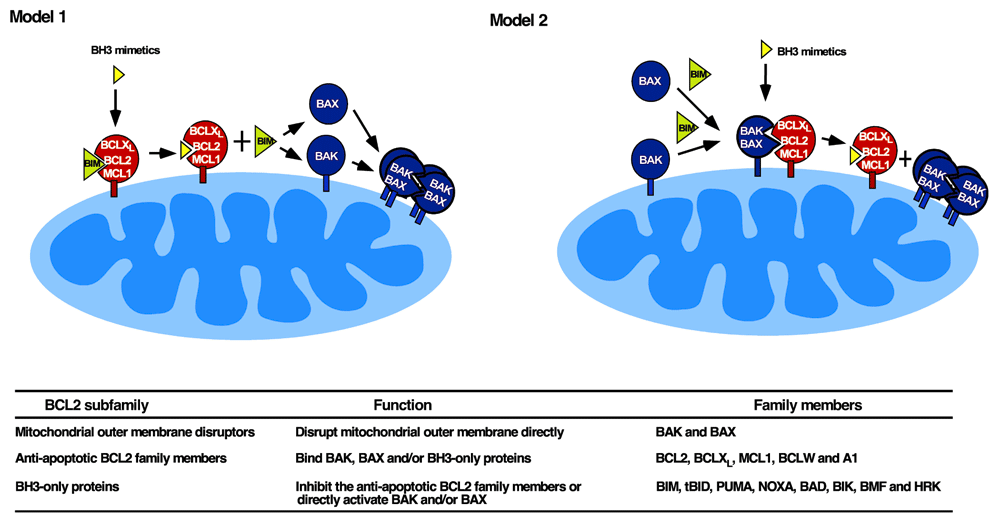
Figure 1. Two models of BH3 mimetic action.
In Model 1 (left), BH3 mimetics are thought to displace activated BIM from anti-apoptotic BCL2 family members, allowing BIM to subsequently activate BAX and BAK44. In Model 2 (right), BAK and/or BAX are constitutively activated and are displaced from anti-apoptotic BCL2 family members by BH3 mimetics46. Model 2 is more compatible with recent studies showing that BAK and BAX can be activated in the absence of BH3-only proteins under cell-free conditions46 and in gene-targeted HCT116 cells123. It is, however, possible that one mode of activation predominates in some cell lines or tissues and the other mode predominates in others. Table below figure, summary of BCL2 subfamilies, their functions, and members.
First, a subset of BH3-only proteins, termed direct activators, can directly activate BAX and BAK. This group of proteins includes BIM, tBID (a cleaved form of BID), and PUMA31,32,38. The role of NOXA as a direct activator has been controversial, with some studies showing activation of BAX or BAK by NOXA protein33,39 and other studies reporting that NOXA BH3 peptide cannot directly activate BAX or BAK40–42. Chen et al. recently reported that interruption of the gene encoding NOXA in cells already lacking BID, BIM, and PUMA causes increased resistance to multiple apoptotic stimuli, suggesting an important role for NOXA in BAX/BAK activation43. To the extent that BH3-only proteins are constitutively activated but sequestered by anti-apoptotic BCL2 family members44,45, displacement of BH3-only proteins by BH3 mimetics can provide a driving force for BAX and BAK activation (Figure 1, Model 1).
Recent results, however, suggest that an alternative mechanism might also contribute to BH3 mimetic-induced killing. In particular, Chen and coworkers reported that BID/BIM/PUMA/NOXA-deficient cells can still undergo apoptosis after certain treatments such as etoposide or ultraviolet light, suggesting the existence of additional BAX/BAK activation pathways43. We simultaneously reported that BAK can undergo lipid-dependent autoactivation under cell-free conditions46. Within intact cells, the extent of constitutive BAK oligomerization (indicative of partial activation) correlated with BAK protein levels across a panel of lymphohematopoietic cell lines. Moreover, BAK knockdown diminished the extent of BAK oligomerization, suggesting concentration-dependent autoactivation in situ46.
If BAK undergoes concentration-dependent autoactivation, how can cells with high BAK levels survive? Our further studies demonstrated that constitutively activated BAK is bound to BCLXL, MCL1, or less commonly BCL246. Based on these observations, BH3 mimetics might be killing cells by displacing partially activated BAK from anti-apoptotic BCL2 family members (Figure 1, Model 2). Consistent with this possibility, cells with constitutive BAK·MCL1 complexes were particularly sensitive to the MCL1 antagonist A-1210477, whereas those with constitutive BAK·BCLXL complexes were more sensitive to the BCL2/BCLXL inhibitor navitoclax46. In agreement with these observations, mice harboring a BAK mutant with reduced affinity for BCLXL had diminished T cells and platelets, suggesting that BCLXL-mediated neutralization of constitutively activated BAK is also important for the survival of certain cell lineages in vivo47. In other cells, e.g. human embryonic stem cells, BAX is constitutively activated, likewise conferring sensitivity to BH3 mimetics48.
Current status of BH3 mimetics
As these complex and highly dynamic interactions between various BCL2 family members were being elucidated, small molecules that could mimic the effects of BH3-only proteins were also being developed. ABT-737, the first compound identified as a bona fide BH3 mimetic8, exhibited high affinity for BCL2, BCLXL, and BCLW (Kd <1 nM) and lower affinity for MCL1 and A1/BFL1 (Kd >1000 nM). Although ABT-737 killed cells in a BAX/BAK-dependent manner49,50 and exhibited anti-tumor activity8, it was not orally bioavailable and displayed poor aqueous solubility, precluding its clinical development.
Navitoclax: inhibitor of BCL2, BCLXL, and BCLW
Navitoclax (ABT-263), an orally bioavailable small molecule with a binding profile similar to that of ABT-7379, also disrupts interactions involving BCL2 and BCLXL, causes BAX/BAK-dependent apoptosis in vitro, and induces complete regressions in xenograft models of small-cell lung cancer (SCLC) and acute lymphoblastic leukemia (ALL)9. In early clinical testing, navitoclax displayed single-agent activity against relapsed/refractory lymphoid malignancies10,51, especially CLL. Adding navitoclax to the monoclonal anti-CD20 antibody rituximab improved both response rate and progression-free survival in previously untreated CLL compared to rituximab alone52. This hypersensitivity of CLL was thought to reflect frequent deletion of genes on chromosome 13q14 encoding miR15A and miR16A, two microRNAs that normally inhibit BCL2 expression53,54. Loss of these microRNAs is thought to result in constitutive BCL2 overexpression and BCL2 addiction5,6,55.
Unfortunately, navitoclax also acutely induced thrombocytopenia10,51, reflecting the role of BCLXL in platelet survival47,56,57. Although this thrombocytopenia could be diminished by treating patients with 150 mg navitoclax/day for one week followed by therapeutic does of 325 mg daily10, maximal BCL2 inhibition was never achieved in lymphoid malignancies because of toxicities of BCLXL inhibition in other normal tissues. Moreover, clinical activity of navitoclax in solid tumors was limited. SCLC appeared to respond better than did other tumors58, but only 3% (one in 39) of patients59 achieved even a partial response (PR). In addition, when navitoclax was combined with other agents, including carboplatin/paclitaxel60, gemcitabine61, or irinotecan62, extensive toxicity and limited efficacy were observed.
Venetoclax: a BCL2-selective inhibitor for CLL and beyond
Developed specifically to avoid the thrombocytopenia associated with BCLXL inhibition, venetoclax exhibits selectivity for BCL2 over BCLXL (Kd <0.01 nM versus 48 nM, respectively), kills cells in a BAX/BAK-dependent manner, and spares platelets11. In light of the navitoclax clinical results, the first phase I trial of venetoclax was conducted in relapsed or refractory (R/R) CLL, including CLL with deletions of the short arm of chromosome 17 (17p), where the tumor suppressor gene TP53 is located, unmutated IGHV, or fludarabine-resistant disease63. Among 116 patients treated, 59% achieved PR and 20% clinical complete remission (CR), including 5% who had no detectable residual disease by flow cytometry. A subsequent single-arm phase II trial demonstrated a 72% PR and 7.5% CR rate in R/R CLL with 17p deletion64. The major side effect in these trials was tumor lysis syndrome (TLS), which could be minimized by starting at a dose of 20 mg daily and ramping up weekly to 400 mg daily over 5 weeks. These observations led to FDA approval of venetoclax for 17p-deleted CLL in April 2016 (http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm495351.htm). However, consistent with the idea that venetoclax, as a BH3 mimetic, should induce apoptosis in a TP53-independent manner, venetoclax kills CLL cells ex vivo regardless of their TP53 mutation status65. Accordingly, a retrospective analysis of TP53 status in cases treated in the original phase I trial63 might clarify whether TP53 wild-type CLL also responds clinically, which could broaden the indication for venetoclax.
Beyond CLL, venetoclax exhibits activity against a variety of lymphoid malignancies. In preclinical studies, concurrent inhibition of BCLXL is required for venetoclax to kill most ALL cells66, the notable exception being MLL-rearranged (MLLr) ALL. In this latter disease, BCL2 is highly expressed because of DOT1L-mediated H3K79 methylation67, rendering MLLr ALL sensitive to venetoclax alone66,67. A clinical trial of venetoclax in MLLr ALL is awaited with interest.
Venetoclax is also active against lymphomas. In a phase I trial, venetoclax monotherapy had an overall response rate of 44% (Table 1) in various R/R non-Hodgkin lymphomas68. Addition of the alkylating agent bendamustine and anti-CD20 antibody rituximab resulted in an even more impressive overall response rate in follicular lymphoma, diffuse large B-cell lymphoma, and marginal zone lymphoma (Table 2)69. In combination with the Bruton’s tyrosine kinase inhibitor ibrutinib, venetoclax also induced remissions in R/R mantle cell lymphoma70.
Table 1. Efficacy of venetoclax monotherapy in relapsed/refractory NHLa.
| Disease | Number | ORb | CR | PR | Stable | PROG | Median progression-
free survival (months) | 12-month
survival |
|---|
| WM | 4 | 100% | 0% | 100% | 0% | 0% | NR | NR |
| MCL | 28 | 75% | 21% | 54% | 18% | 4% | 14 | 82% |
| MZL | 3 | 67% | 0% | 67% | 0% | 0% | NR | NR |
| DLBCL-RT | 7 | 43% | 0% | 43% | 29% | 14% | NR | NR |
| DLBCL | 34 | 18% | 12% | 6% | 24% | 56% | 1 | 34% |
| FL | 29 | 38% | 14% | 24% | 59% | 4% | 11 | 100% |
| Total | 106 | 44% | 13% | 31% | 30% | 22% | 17 | 72% |
Table 2. Efficacy of bendamustine/rituximab/venetoclax against NHLa.
| FLb | DLBCL | MZL | |
|---|
| Number of patients | 27 | 16 | 6 | |
| OR | 78% | 38% | 80% | |
| CR | 30% | 25% | 20% | |
| PR | 48% | 13% | 60% | |
| Stable | 4% | 13% | 0% | |
| PROG | 7% | 38% | 0% | |
BCL2 has also been implicated in the survival of multiple myeloma (MM) cells, particularly those with t(11;14) translocation. Accordingly, MMs with this translocation have a higher response rate to venetoclax than those without (24% versus 4%)71. In addition, a trial of venetoclax in combination with bortezomib and dexamethasone in R/R MM appears promising72.
Venetoclax has also been extensively studied in acute myelogenous leukemia (AML). A preclinical study suggested that AML is exquisitely sensitive to single-agent venetoclax ex vivo73. A subsequent phase II clinical trial, however, demonstrated responses (CR/CRi) in only six of 32 patients (19%) with R/R AML74. This somewhat low response rate may be related to the upregulation of BCLXL and MCL1 in this disease, particularly at the time of AML relapse75, as well as other factors such as HOX gene expression76. Interestingly, combinations of venetoclax with low-dose cytarabine or DNA methyltransferase inhibitors exhibit response rates of 44%77 and 76%78, respectively, in elderly patients with previously untreated AML, raising the possibility that using venetoclax as a sensitizing agent might be particularly effective in this patient population.
In most solid tumors, BCLXL and MCL1 appear to be more important than BCL2 in inhibiting apoptosis79. However, SCLC80 and estrogen receptor-positive (ER+) breast cancer81 exhibit high BCL2 expression and venetoclax sensitivity. Accordingly, clinical studies of venetoclax in these malignancies appear to be warranted but have not yet been initiated.
BCLXL inhibitor: WEHI-539
BCLXL is frequently expressed at high levels in solid tumors, including colorectal82, hormone-refractory prostate83, and mesenchymal breast cancers84, conferring chemotherapy resistance84,85. A BCLXL inhibitor could be particularly useful in treating these cancers. WEHI-53986 and its more potent derivative A-115546387 selectively and tightly bind to BCLXL. In mice, A-1155463 causes reversible on-target thrombocytopenia and inhibits SCLC xenograft growth87. In addition, WEHI-539 synergizes with carboplatin in ovarian cancer cell lines88 and with doxorubicin in osteosarcoma89.
Because of the role of BCLXL in platelet survival47,56,57, clinical application of a selective BCLXL inhibitor could be a challenge. As with navitoclax, a possible lead-in dose of BCLXL inhibitor may lower the risk of severe thrombocytopenia, but clinical efficacy might also be compromised because suboptimal BCLXL inhibition during the lead-in might facilitate the development of tumor cell tolerance. Therefore, development of an alternative strategy to mitigate thrombocytopenia might be important for the successful application of BCLXL inhibitors.
MCL1 inhibitor: A-1210477
MCL1 is also an attractive target. MCL1 elevation occurs in many tumors75,79,90,91 and is associated with poor prognosis92–98. Moreover, MCL1 contributes to therapy resistance99, especially to ABT-737, navitoclax, and venetoclax49,100–103.
To date, only one MCL1-selective inhibitor, A1210477, has been reported102. This agent disrupts complexes of MCL1 with BH3-only proteins, kills MCL1-dependent cells, and exhibits synergy with navitoclax in vitro102,104. Studying an agent with these properties in vivo could potentially be productive.
Prediction of BH3 mimetic sensitivity or resistance
Even though venetoclax has substantial activity against 17p-deleted CLL, responses are not universal. In an era of increasing emphasis on precision medicine, there is substantial interest in predicting which cases will respond to venetoclax or other BH3 mimetics and which will not.
BCL2 family protein levels
Several groups have reported that chemotherapy sensitivity can be predicted by algorithms that essentially measure levels of anti-apoptotic BCL2 family members, sum the values, and subtract levels of BAX and BAK105–107. While this approach detects differences between sensitive and resistant groups of cell lines or tumors, overlap between the groups might make it difficult to use this approach to dictate therapy for individual patients. Moreover, this approach generally fails to take into account endogenous levels of BH3-only proteins and other binding partners that could alter the anti-apoptotic or pro-apoptotic potentials of the proteins assayed.
For assessing venetoclax sensitivity of lymphoid malignancies, the calculation might actually be simpler. Myc-transformed murine lymphomas are sensitive to navitoclax only if they have elevated BCL2 levels107. Accordingly, measurement of BCL2 alone might help predict sensitive versus resistant CLL cases. Consistent with this possibility, recent studies have reported that high BCL2 expression correlates with venetoclax sensitivity in neoplastic lymphocytes11,108. Whether elevated levels of BCLXL or MCL1, either as a consequence of gene amplification79 or other modifications109,110, will similarly predict sensitivity to selective antagonists of these two proteins remains to be tested.
BH3 profiling assays
BH3 profiling involves treating mitochondria with BH3 peptides and measuring cytochrome c release or mitochondrial depolarization as a strategy to predict sensitivity to BH3 mimetics111,112 or therapies that act through inducing BH3-only proteins113,114. Because the BAD BH3 and HRK BH3 domains have different affinities for anti-apoptotic BCL2 family proteins44,115, with BAD binding BCL2 and BCLXL tightly but HRK binding only BCLXL, subtracting the cytochrome c release caused by HRK from that caused by BAD (BAD–HRK) reportedly predicts venetoclax sensitivity116. Results using this assay suggested that the maturation stage of T-ALLs determines their sensitivity to navitoclax or venetoclax, with most T-ALLs exhibiting navitoclax sensitivity but early T-cell progenitor ALL being sensitive to venetoclax116. This assay also predicted that a substantial percentage of AMLs would be sensitive to venetoclax73. In a subsequent phase II study of venetoclax monotherapy in AML, however, BH3 profiling results correlated only weakly with time on study74, suggesting that determinants of response are more complicated than originally envisioned.
Building on the experience with BH3 profiling, a modified assay called “dynamic BH3 profiling” involves exposure of cells to diluent versus any potential anticancer drug or combination followed by assessment of mitochondrial depolarization by BIM BH3 peptide in permeabilized cells117. Early experience with this assay in multiple model systems indicates that drug-induced increases in BIM BH3 peptide-induced mitochondrial depolarization after 16 hours of drug exposure correlate with the extent of cell death at 72–96 hours of continuous drug exposure ex vivo. Whether this assay will provide improved ability to predict response to BH3 mimetics in the clinical setting remains to be determined.
Preformed complexes as potential predictors of response
An alternative approach to predicting BH3 mimetic sensitivity might come from recent studies demonstrating constitutive BAK activation in a variety of cells46,47. If BAK is constitutively bound to BCLXL, cells are significantly more sensitive to navitoclax, and if BAK is constitutively bound to MCL1, cells are more sensitive to A-121047746, suggesting that measurement of preformed BCLXL·BAK and MCL1·BAK complexes might provide insight into sensitivity to the respective BH3 mimetics. There is also a correlation between preformed BCL2·BAK complexes and venetoclax sensitivity46, perhaps reflecting the fact that these complexes, though somewhat less stable, nonetheless form when BCL2 is expressed at high levels or harbors gain-of-function mutations118,119. All of these complexes between BAK and anti-apoptotic BCL2 family members can, like complexes between BH3-only proteins and anti-apoptotic BCL2 family members44,45, be detected and potentially quantified by immunoprecipitation46. Because anti-apoptotic BCL2 family members are expressed on the cytoplasmic surfaces of multiple organelles, not just mitochondria120,121, it is possible that immunoprecipitation followed by immunoblotting for BAK, BAX, and BH3-only proteins will provide a more complete picture of the cellular balance between pro-apoptotic and anti-apoptotic BCL2 family members than MOM permeabilization analyses alone.
Opportunities for future development
While the recent FDA approval of venetoclax marks a milestone in apoptosis research, there is still much work to be done. As mentioned above, the results of single-agent venetoclax trials in TP53 wild-type CLL, other lymphoid malignancies, SCLC, and ER+ breast cancer are awaited with interest. Moreover, further studies examining the optimal use of venetoclax as a chemosensitizing agent are needed.
Based on the observed clinical activity of venetoclax in CLL, the prospect of selectively targeting BCLXL and MCL1, especially in cancers with BCLX or MCL1 amplification79, is also appealing. There are, however, substantial obstacles. The role of BCLXL in platelet survival and the consequent thrombocytopenia induced by BCLXL inhibition hampered the development of navitoclax10. Whether it will be possible to develop a clinically viable strategy for avoiding or overcoming this on-target side effect with selective BCLXL inhibitors remains to be determined. Likewise, it was reported over a decade ago that MCL1 is required for hematopoietic stem cell survival122. Because it now appears that this might be due to a BH3-binding groove-independent role for MCL1 in oxidative phosphorylation106 rather than the role of MCL1 in apoptosis, it is possible that MCL1-selective BH3 mimetics will not be as toxic to normal cells as MCL1 gene disruption. The development of an MCL1 inhibitor that can be applied in preclinical tumor models and possibly in clinical trials would allow this hypothesis to be tested.
Finally, a substantial fraction of tumors might be resistant to selective BCL2, BCLXL, or MCL1 inhibitors. To the extent that these agents act by releasing partially activated BAK or BAX from pre-formed complexes46, cells lacking activated BAK and BAX will experience little or no effect from these inhibitors. One strategy for sensitizing these cells to BH3 mimetics would be to treat with chemotherapeutic agents that activate BH3-only proteins, leading to BAK or BAX activation46. Alternatively, it might be possible to induce apoptosis in these cells using BH3 mimetics that directly activate BAK and/or BAX. Whether it will be possible to derive such compounds and target them in a way that allows cancer cell-selective killing also remains to be determined.
Competing interests
The authors declare that they have no competing interests.
Grant information
Preparation of this review was supported in part by a grant from the NIH (R01 CA166741). Haiming Dai is also supported by the Hundred-Talent Program of the Chinese Academy of Sciences.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We thank members of the Kaufmann lab for provocative discussions and Deb Strauss for editorial assistance. We also apologize to many colleagues whose important contributions could not be highlighted because of space limitations.
Faculty Opinions recommendedReferences
- 1.
Tsujimoto Y, Cossman J, Jaffe E, et al.:
Involvement of the bcl-2 gene in human follicular lymphoma.
Science.
1985; 228(4706): 1440–3. PubMed Abstract
| Publisher Full Text
- 2.
Cleary ML, Smith SD, Sklar J:
Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation.
Cell.
1986; 47(1): 19–28. PubMed Abstract
| Publisher Full Text
- 3.
Vaux DL, Cory S, Adams JM:
Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells.
Nature.
1988; 335(6189): 440–2. PubMed Abstract
| Publisher Full Text
- 4.
Strasser A, Harris AW, Cory S:
bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship.
Cell.
1991; 67(5): 889–99. PubMed Abstract
| Publisher Full Text
- 5.
Hanada M, Delia D, Aiello A, et al.:
bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia.
Blood.
1993; 82(6): 1820–8. PubMed Abstract
- 6.
Robertson LE, Plunkett W, McConnell K, et al.:
Bcl-2 expression in chronic lymphocytic leukemia and its correlation with the induction of apoptosis and clinical outcome.
Leukemia.
1996; 10(3): 456–9. PubMed Abstract
- 7.
Sattler M, Liang H, Nettesheim D, et al.:
Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis.
Science.
1997; 275(5302): 983–6. PubMed Abstract
| Publisher Full Text
- 8.
Oltersdorf T, Elmore SW, Shoemaker AR, et al.:
An inhibitor of Bcl-2 family proteins induces regression of solid tumours.
Nature.
2005; 435(7042): 677–81. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 9.
Tse C, Shoemaker AR, Adickes J, et al.:
ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor.
Cancer Res.
2008; 68(9): 3421–8. PubMed Abstract
| Publisher Full Text
- 10.
Roberts AW, Seymour JF, Brown JR, et al.:
Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease.
J Clin Oncol.
2012; 30(5): 488–96. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 11.
Souers AJ, Leverson JD, Boghaert ER, et al.:
ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets.
Nat Med.
2013; 19(2): 202–8. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 12.
Ni Chonghaile T, Letai A:
Mimicking the BH3 domain to kill cancer cells.
Oncogene.
2008; 27(Suppl 1): S149–57. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 13.
Strasser A, Cory S, Adams JM:
Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases.
EMBO J.
2011; 30(18): 3667–83. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 14.
Anderson MA, Huang D, Roberts A:
Targeting BCL2 for the treatment of lymphoid malignancies.
Semin Hematol.
2014; 51(3): 219–27. PubMed Abstract
| Publisher Full Text
- 15.
Martinou JC, Youle RJ:
Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics.
Dev Cell.
2011; 21(1): 92–101. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 16.
Fuchs Y, Steller H:
Programmed cell death in animal development and disease.
Cell.
2011; 147(4): 742–58. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 17.
Hyman BT, Yuan J:
Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology.
Nat Rev Neurosci.
2012; 13(6): 395–406. PubMed Abstract
| Publisher Full Text
- 18.
Jiang X, Wang X:
Cytochrome C-mediated apoptosis.
Annu Rev Biochem.
2004; 73: 87–106. PubMed Abstract
| Publisher Full Text
- 19.
Ekert PG, Vaux DL:
The mitochondrial death squad: hardened killers or innocent bystanders?
Curr Opin Cell Biol.
2005; 17(6): 626–30. PubMed Abstract
| Publisher Full Text
- 20.
Taylor RC, Cullen SP, Martin SJ:
Apoptosis: controlled demolition at the cellular level.
Nat Rev Mol Cell Biol.
2008; 9(3): 231–41. PubMed Abstract
| Publisher Full Text
- 21.
Cory S, Adams JM:
The Bcl2 family: regulators of the cellular life-or-death switch.
Nat Rev Cancer.
2002; 2(9): 647–56. PubMed Abstract
| Publisher Full Text
- 22.
Czabotar PE, Lessene G, Strasser A, et al.:
Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy.
Nat Rev Mol Cell Biol.
2014; 15(1): 49–63. PubMed Abstract
| Publisher Full Text
- 23.
Antonsson B, Conti F, Ciavatta A, et al.:
Inhibition of Bax channel-forming activity by Bcl-2.
Science.
1997; 277(5324): 370–2. PubMed Abstract
| Publisher Full Text
- 24.
Ma S, Hockings C, Anwari K, et al.:
Assembly of the Bak apoptotic pore: a critical role for the Bak protein α6 helix in the multimerization of homodimers during apoptosis.
J Biol Chem.
2013; 288(36): 26027–38. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 25.
Salvador-Gallego R, Mund M, Cosentino K, et al.:
Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores.
EMBO J.
2016; 35(4): 389–401. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 26.
Große L, Wurm CA, Brüser C, et al.:
Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis.
EMBO J.
2016; 35(4): 402–13. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 27.
Basañez G, Nechushtan A, Drozhinin O, et al.:
Bax, but not Bcl-xL, decreases the lifetime of planar phospholipid bilayer membranes at subnanomolar concentrations.
Proc Natl Acad Sci U S A.
1999; 96(10): 5492–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 28.
Terrones O, Etxebarria A, Landajuela A, et al.:
BIM and tBID are not mechanistically equivalent when assisting BAX to permeabilize bilayer membranes.
J Biol Chem.
2008; 283(12): 7790–803. PubMed Abstract
| Publisher Full Text
- 29.
Westphal D, Dewson G, Menard M, et al.:
Apoptotic pore formation is associated with in-plane insertion of Bak or Bax central helices into the mitochondrial outer membrane.
Proc Natl Acad Sci U S A.
2014; 111(39): E4076–85. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 30.
Kuwana T, Olson NH, Kiosses WB, et al.:
Pro-apoptotic Bax molecules densely populate the edges of membrane pores.
Sci Rep.
2016; 6: 27299. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 31.
Letai A, Bassik MC, Walensky LD, et al.:
Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics.
Cancer Cell.
2002; 2(3): 183–92. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 32.
Kuwana T, Bouchier-Hayes L, Chipuk JE, et al.:
BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly.
Mol Cell.
2005; 17(4): 525–35. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 33.
Dai H, Smith A, Meng XW, et al.:
Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization.
J Cell Biol.
2011; 194(1): 39–48. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 34.
Willis SN, Fletcher JI, Kaufmann T, et al.:
Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak.
Science.
2007; 315(5813): 856–9. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 35.
Llambi F, Moldoveanu T, Tait SW, et al.:
A unified model of mammalian BCL-2 protein family interactions at the mitochondria.
Mol Cell.
2011; 44(4): 517–31. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 36.
Muchmore SW, Sattler M, Liang H, et al.:
X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death.
Nature.
1996; 381(6580): 335–41. PubMed Abstract
| Publisher Full Text
- 37.
Petros AM, Medek A, Nettesheim DG, et al.:
Solution structure of the antiapoptotic protein bcl-2.
Proc Natl Acad Sci U S A.
2001; 98(6): 3012–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 38.
Dai H, Pang Y, Ramirez-Alvarado M, et al.:
Evaluation of the BH3-only protein Puma as a direct Bak activator.
J Biol Chem.
2014; 289(1): 89–99. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 39.
Du H, Wolf J, Schafer B, et al.:
BH3 domains other than Bim and Bid can directly activate Bax/Bak.
J Biol Chem.
2011; 286(1): 491–501. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 40.
Moldoveanu T, Grace CR, Llambi F, et al.:
BID-induced structural changes in BAK promote apoptosis.
Nat Struct Mol Biol.
2013; 20(5): 589–97. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 41.
Czabotar PE, Westphal D, Dewson G, et al.:
Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis.
Cell.
2013; 152(3): 519–31. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 42.
Brouwer JM, Westphal D, Dewson G, et al.:
Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers.
Mol Cell.
2014; 55(6): 938–46. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 43.
Chen HC, Kanai M, Inoue-Yamauchi A, et al.:
An interconnected hierarchical model of cell death regulation by the BCL-2 family.
Nat Cell Biol.
2015; 17(10): 1270–81. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 44.
Certo M, Del Gaizo Moore V, Nishino M, et al.:
Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members.
Cancer Cell.
2006; 9(5): 351–65. PubMed Abstract
| Publisher Full Text
- 45.
Dai H, Ding H, Meng XW, et al.:
Contribution of Bcl-2 phosphorylation to Bak binding and drug resistance.
Cancer Res.
2013; 73(23): 6998–7008. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 46.
Dai H, Ding H, Meng XW, et al.:
Constitutive BAK activation as a determinant of drug sensitivity in malignant lymphohematopoietic cells.
Genes Dev.
2015; 29(20): 2140–52. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 47.
Lee EF, Grabow S, Chappaz S, et al.:
Physiological restraint of Bak by Bcl-xL is essential for cell survival.
Genes Dev.
2016; 30(10): 1240–50. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 48.
Dumitru R, Gama V, Fagan BM, et al.:
Human embryonic stem cells have constitutively active Bax at the Golgi and are primed to undergo rapid apoptosis.
Mol Cell.
2012; 46(5): 573–83. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 49.
van Delft MF, Wei AH, Mason KD, et al.:
The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized.
Cancer Cell.
2006; 10(5): 389–99. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 50.
Vogler M, Weber K, Dinsdale D, et al.:
Different forms of cell death induced by putative BCL2 inhibitors.
Cell Death Differ.
2009; 16(7): 1030–9. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 51.
Chiappori AA, Schreeder MT, Moezi MM, et al.:
A phase I trial of pan-Bcl-2 antagonist obatoclax administered as a 3-h or a 24-h infusion in combination with carboplatin and etoposide in patients with extensive-stage small cell lung cancer.
Br J Cancer.
2012; 106(5): 839–45. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 52.
Kipps TJ, Eradat H, Grosicki S, et al.:
A phase 2 study of the BH3 mimetic BCL2 inhibitor navitoclax (ABT-263) with or without rituximab, in previously untreated B-cell chronic lymphocytic leukemia.
Leuk Lymphoma.
2015; 56(10): 2826–33. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 53.
Cimmino A, Calin GA, Fabbri M, et al.:
miR-15 and miR-16 induce apoptosis by targeting BCL2.
Proc Natl Acad Sci U S A.
2005; 102(39): 13944–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 54.
Klein U, Lia M, Crespo M, et al.:
The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia.
Cancer Cell.
2010; 17(1): 28–40. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 55.
Del Gaizo Moore V, Brown JR, Certo M, et al.:
Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737.
J Clin Invest.
2007; 117(1): 112–21. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 56.
Mason KD, Carpinelli MR, Fletcher JI, et al.:
Programmed anuclear cell death delimits platelet life span.
Cell.
2007; 128(6): 1173–86. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 57.
Josefsson EC, White MJ, Dowling MR, et al.:
Platelet life span and apoptosis.
Methods Mol Biol.
2012; 788: 59–71. PubMed Abstract
| Publisher Full Text
- 58.
Gandhi L, Camidge DR, Ribeiro de Oliveira M, et al.:
Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors.
J Clin Oncol.
2011; 29(7): 909–16. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 59.
Rudin CM, Hann CL, Garon EB, et al.:
Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer.
Clin Cancer Res.
2012; 18(11): 3163–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 60.
Vlahovic G, Karantza V, Wang D, et al.:
A phase I safety and pharmacokinetic study of ABT-263 in combination with carboplatin/paclitaxel in the treatment of patients with solid tumors.
Invest New Drugs.
2014; 32(5): 976–84. PubMed Abstract
| Publisher Full Text
- 61.
Cleary JM, Lima CM, Hurwitz HI, et al.:
A phase I clinical trial of navitoclax, a targeted high-affinity Bcl-2 family inhibitor, in combination with gemcitabine in patients with solid tumors.
Invest New Drugs.
2014; 32(5): 937–45. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 62.
Tolcher AW, LoRusso P, Arzt J, et al.:
Safety, efficacy, and pharmacokinetics of navitoclax (ABT-263) in combination with irinotecan: results of an open-label, phase 1 study.
Cancer Chemother Pharmacol.
2015; 76(5): 1041–9. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 63.
Roberts AW, Davids MS, Pagel JM, et al.:
Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia.
N Engl J Med.
2016; 374(4): 311–22. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 64.
Stilgenbauer S, Eichhorst B, Schetelig J, et al.:
Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study.
Lancet Oncol.
2016; 17(6): 768–78. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 65.
Anderson MA, Deng J, Seymour JF, et al.:
The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism.
Blood.
2016; 127(25): 3215–24. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 66.
Khaw SL, Suryani S, Evans K, et al.:
Venetoclax responses of pediatric ALL xenografts reveal sensitivity of MLL-rearranged leukemia.
Blood.
2016; 128(10): 1382–95. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 67.
Benito JM, Godfrey L, Kojima K, et al.:
MLL-Rearranged Acute Lymphoblastic Leukemias Activate BCL-2 through H3K79 Methylation and Are Sensitive to the BCL-2-Specific Antagonist ABT-199.
Cell Rep.
2015; 13(12): 2715–27. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 68.
Gerecitano JF, Roberts AW, Seymour JF, et al.:
A Phase 1 Study of Venetoclax (ABT-199 / GDC-0199) Monotherapy in Patients with Relapsed/Refractory Non-Hodgkin Lymphoma.
Blood.
2015; 126(23): 254. Reference Source
- 69.
de Vos S, Swinnen L, Kozloff M, et al.:
A Dose-Escalation Study of Venetoclax (ABT-199/GDC-0199) in Combination with Bendamustine and Rituximab in Patients with Relapsed or Refractory Non-Hodgkin's Lymphoma.
Blood.
2015; 126(23): 255. Reference Source
- 70.
Tam CSL, Roberts AW, Anderson MA, et al.:
The combination of ibrutinib and venetoclax (ABT-199) to achieve complete remissions in patients with relapsed/refractory mantle cell lymphoma: Preliminary results of the phase II AIM study.
J Clin Oncol.
2016; 34(suppl): absract 7548. Reference Source
- 71.
Kumar S, Vij R, Kaufman JL, et al.:
Phase I venetoclax monotherapy for relapsed/refractory multiple myeloma.
J Clin Oncol.
2016; 34(suppl): abstract 8032. Reference Source
- 72.
Moreau P, Chanan-Khan AAA, Roberts AW, et al.:
Phase Ib venetoclax combined with bortezomib and dexamethasone in relapsed/refractory multiple myeloma.
J Clin Oncol.
2016; 34(suppl): abstract 8011. Reference Source
- 73.
Pan R, Hogdal LJ, Benito JM, et al.:
Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia.
Cancer Discov.
2014; 4(3): 362–75. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 74.
Konopleva M, Pollyea DA, Potluri J, et al.:
Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia.
Cancer Discov.
2016; 6(10): 1106–17. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 75.
Kaufmann SH, Karp JE, Svingen PA, et al.:
Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse.
Blood.
1998; 91(3): 991–1000. PubMed Abstract
- 76.
Kontro M, Kumar A, Majumder MM, et al.:
HOX gene expression predicts response to BCL-2 inhibition in acute myeloid leukemia.
Leukemia.
2016. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 77.
Lin TL, Strickland SA, Fiedler W, et al.:
Phase Ib/2 study of venetoclax with low-dose cytarabine in treatment-naive patients age ≥ 65 with acute myelogenous leukemia.
J Clin Oncol.
2016; 34(suppl): abstract 7007. Reference Source
- 78.
Pollyea DA, Dinardo CD, Thirman MJ, et al.:
Results of a phase 1b study of venetoclax plus decitabine or azacitidine in untreated acute myeloid leukemia patients ≥ 65 years ineligible for standard induction therapy.
J Clin Oncol.
2015; 34(Suppl): (suppl., abstract 7009). Reference Source
- 79.
Beroukhim R, Mermel CH, Porter D, et al.:
The landscape of somatic copy-number alteration across human cancers.
Nature.
2010; 463(7283): 899–905. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 80.
Jiang SX, Sato Y, Kuwao S, et al.:
Expression of bcl-2 oncogene protein is prevalent in small cell lung carcinomas.
J Pathol.
1995; 177(2): 135–8. PubMed Abstract
| Publisher Full Text
- 81.
Vaillant F, Merino D, Lee L, et al.:
Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen receptor-positive breast cancer.
Cancer Cell.
2013; 24(1): 120–9. PubMed Abstract
| Publisher Full Text
- 82.
Zhang H, Xue J, Hessler P, et al.:
Genomic analysis and selective small molecule inhibition identifies BCL-XL as a critical survival factor in a subset of colorectal cancer.
Mol Cancer.
2015; 14: 126. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 83.
Castilla C, Congregado B, Chinchón D, et al.:
Bcl-xL is overexpressed in hormone-resistant prostate cancer and promotes survival of LNCaP cells via interaction with proapoptotic Bak.
Endocrinology.
2006; 147(10): 4960–7. PubMed Abstract
| Publisher Full Text
- 84.
Keitel U, Scheel A, Thomale J, et al.:
Bcl-xL mediates therapeutic resistance of a mesenchymal breast cancer cell subpopulation.
Oncotarget.
2014; 5(23): 11778–91. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 85.
Amundson SA, Myers TG, Scudiero D, et al.:
An informatics approach identifying markers of chemosensitivity in human cancer cell lines.
Cancer Res.
2000; 60(21): 6101–10. PubMed Abstract
- 86.
Lessene G, Czabotar PE, Sleebs BE, et al.:
Structure-guided design of a selective BCL-XL inhibitor.
Nat Chem Biol.
2013; 9(6): 390–7. PubMed Abstract
| Publisher Full Text
- 87.
Tao ZF, Hasvold L, Wang L, et al.:
Discovery of a Potent and Selective BCL-XL Inhibitor with in Vivo Activity.
ACS Med Chem Lett.
2014; 5(10): 1088–93. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 88.
Abed MN, Abdullah MI, Richardson A:
Antagonism of Bcl-XL is necessary for synergy between carboplatin and BH3 mimetics in ovarian cancer cells.
J Ovarian Res.
2016; 9: 25. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 89.
Baranski Z, de Jong Y, Ilkova T, et al.:
Pharmacological inhibition of Bcl-xL sensitizes osteosarcoma to doxorubicin.
Oncotarget.
2015; 6(34): 36113–25. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 90.
Hussain SR , Cheney CM, Johnson AJ, et al.:
Mcl-1 is a relevant therapeutic target in acute and chronic lymphoid malignancies: down-regulation enhances rituximab-mediated apoptosis and complement-dependent cytotoxicity.
Clin Cancer Res.
2007; 13(7): 2144–50. PubMed Abstract
| Publisher Full Text
- 91.
Kitada S, Andersen J, Akar S, et al.:
Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with In vitro and In vivo chemoresponses.
Blood.
1998; 91(9): 3379–89. PubMed Abstract
- 92.
Backus HH, van Riel JM, van Groeningen CJ, et al.:
Rb, mcl-1 and p53 expression correlate with clinical outcome in patients with liver metastases from colorectal cancer.
Ann Oncol.
2001; 12(6): 779–85. PubMed Abstract
| Publisher Full Text
- 93.
Shigemasa K, Katoh O, Shiroyama Y, et al.:
Increased MCL-1 expression is associated with poor prognosis in ovarian carcinomas.
Jpn J Cancer Res.
2002; 93(5): 542–50. PubMed Abstract
| Publisher Full Text
- 94.
Wuillème-Toumi S, Robillard N, Gomez P, et al.:
Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival.
Leukemia.
2005; 19(7): 1248–52. PubMed Abstract
| Publisher Full Text
- 95.
Sieghart W, Losert D, Strommer S, et al.:
Mcl-1 overexpression in hepatocellular carcinoma: a potential target for antisense therapy.
J Hepatol.
2006; 44(1): 151–7. PubMed Abstract
| Publisher Full Text
- 96.
Zhuang L, Lee CS, Scolyer RA, et al.:
Mcl-1, Bcl-XL and Stat3 expression are associated with progression of melanoma whereas Bcl-2, AP-2 and MITF levels decrease during progression of melanoma.
Mod Pathol.
2007; 20(4): 416–26. PubMed Abstract
| Publisher Full Text
- 97.
Ding Q, He X, Xia W, et al.:
Myeloid cell leukemia-1 inversely correlates with glycogen synthase kinase-3beta activity and associates with poor prognosis in human breast cancer.
Cancer Res.
2007; 67(10): 4564–71. PubMed Abstract
| Publisher Full Text
- 98.
Quinn BA, Dash R, Azab B, et al.:
Targeting Mcl-1 for the therapy of cancer.
Expert Opin Investig Drugs.
2011; 20(10): 1397–411. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 99.
Meng XW, Lee SH, Dai H, et al.:
Mcl-1 as a buffer for proapoptotic Bcl-2 family members during TRAIL-induced apoptosis: a mechanistic basis for sorafenib (Bay 43-9006)-induced TRAIL sensitization.
J Biol Chem.
2007; 282(41): 29831–46. PubMed Abstract
| Publisher Full Text
- 100.
Konopleva M, Contractor R, Tsao T, et al.:
Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia.
Cancer Cell.
2006; 10(5): 375–88. PubMed Abstract
| Publisher Full Text
- 101.
Chen S, Dai Y, Harada H, et al.:
Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation.
Cancer Res.
2007; 67(2): 782–91. PubMed Abstract
| Publisher Full Text
- 102.
Leverson JD, Zhang H, Chen J, et al.:
Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax).
Cell Death Dis.
2015; 6: e1590. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 103.
Yecies D, Carlson NE, Deng J, et al.:
Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1.
Blood.
2010; 115(16): 3304–13. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 104.
Xiao Y, Nimmer P, Sheppard GS, et al.:
MCL-1 Is a Key Determinant of Breast Cancer Cell Survival: Validation of MCL-1 Dependency Utilizing a Highly Selective Small Molecule Inhibitor.
Mol Cancer Ther.
2015; 14(8): 1837–47. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 105.
Al-Harbi S, Hill BT, Mazumder S, et al.:
An antiapoptotic BCL-2 family expression index predicts the response of chronic lymphocytic leukemia to ABT-737.
Blood.
2011; 118(13): 3579–90. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 106.
Perciavalle RM, Stewart DP, Koss B, et al.:
Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration.
Nat Cell Biol.
2012; 14(6): 575–83. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 107.
Mérino D, Khaw SL, Glaser SP, et al.:
Bcl-2, Bcl-xL, and Bcl-w are not equivalent targets of ABT-737 and navitoclax (ABT-263) in lymphoid and leukemic cells.
Blood.
2012; 119(24): 5807–16. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 108.
Khaw SL, Mérino D, Anderson MA, et al.:
Both leukaemic and normal peripheral B lymphoid cells are highly sensitive to the selective pharmacological inhibition of prosurvival Bcl-2 with ABT-199.
Leukemia.
2014; 28(6): 1207–15. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 109.
Schwickart M, Huang X, Lill JR, et al.:
Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival.
Nature.
2010; 463(7277): 103–7. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 110.
Wertz IE, Kusam S, Lam C, et al.:
Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7.
Nature.
2011; 471(7336): 110–4. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 111.
Deng J, Carlson N, Takeyama K, et al.:
BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents.
Cancer Cell.
2007; 12(2): 171–85. PubMed Abstract
| Publisher Full Text
- 112.
Ryan JA, Brunelle JK, Letai A:
Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes.
Proc Natl Acad Sci U S A.
2010; 107(29): 12895–900. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 113.
Ni Chonghaile T, Sarosiek KA, Vo TT, et al.:
Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy.
Science.
2011; 334(6059): 1129–33. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 114.
Vo TT, Ryan J, Carrasco R, et al.:
Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML.
Cell.
2012; 151(2): 344–55. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 115.
Chen L, Willis SN, Wei A, et al.:
Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function.
Mol Cell.
2005; 17(3): 393–403. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 116.
Chonghaile TN, Roderick JE, Glenfield C, et al.:
Maturation stage of T-cell acute lymphoblastic leukemia determines BCL-2 versus BCL-XL dependence and sensitivity to ABT-199.
Cancer Discov.
2014; 4(9): 1074–87. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 117.
Montero J, Sarosiek KA, DeAngelo JD, et al.:
Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy.
Cell.
2015; 160(5): 977–89. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 118.
Dai H, Meng XW, Lee SH, et al.:
Context-dependent Bcl-2/Bak interactions regulate lymphoid cell apoptosis.
J Biol Chem.
2009; 284(27): 18311–22. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 119.
Smith AJ, Dai H, Correia C, et al.:
Noxa/Bcl-2 protein interactions contribute to bortezomib resistance in human lymphoid cells.
J Biol Chem.
2011; 286(20): 17682–92. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 120.
Krajewski S, Tanaka S, Takayama S, et al.:
Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes.
Cancer Res.
1993; 53(19): 4701–14. PubMed Abstract
- 121.
Krajewski S, Bodrug S, Gascoyne R, et al.:
Immunohistochemical analysis of Mcl-1 and Bcl-2 proteins in normal and neoplastic lymph nodes.
Am J Pathol.
1994; 145(3): 515–25. PubMed Abstract
| Free Full Text
- 122.
Opferman JT, Iwasaki H, Ong CC, et al.:
Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells.
Science.
2005; 307(5712): 1101–4. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 123.
O'Neill KL, Huang K, Zhang J, et al.:
Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane.
Genes Dev.
2016; 30(8): 973–88. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
Comments on this article Comments (0)