Keywords
Phylogeny, Work-flow, Pipeline, Supermatrix, Supertree
This article is included in the Phylogenetics collection.
Phylogeny, Work-flow, Pipeline, Supermatrix, Supertree
The improvement of high throughput sequencing methods, and the ability to produce more and/or longer reads at a cheaper price have allowed the use of these data in new areas of research (Ellison et al., 2011; Jumpponen & Jones, 2009; Lemmon et al., 2012). More or less, automated software pipelines are often an intrinsic part in the development of ways to process and analyze data for new types of research questions.
Phylogenetic analyses are an integral part of many biological studies. Phylommand is a package of four software - treebender, treeator, contree, and pairalign - with capabilities in manipulation and analyses of phylogenetic trees. The functions include rooting, splitting, and comparing trees, calculating parsimony and likelihood scores, as well as performing parsimonious stepwise addition of taxa, nearest neighbour interchange branch swapping, and construction of neighbour joining trees. In addition there are functions, such as calculating decisiveness (Sanderson et al., 2010), MAD scores (Smith et al., 2009), and matrix representation of trees, for use in supermatrix and supertree pipelines. Similar to the Newick utilities software suit (Junier & Zdobnov, 2010), that also act on phylogenetic trees, the phylommand programs are designed to be used on the command line, without the overhead from a graphical interface. Phylommand also accepts inputs from file or standard input, outputs the results to standard output (i.e. usually screen if not redirected), and works without any configuration files or user input (after execution). It is thus made to work in software pipelines.
Both phylommand and Newick utilities can use the newick file format and are therefore compatible, and they complement each other as most of the functions in phylommand are not included in the Newick utilities. Even if phylommand is made for pipelines, each software work independent of any other program (given appropriate input).
Phylommand is written in the C++ programming language and is primarily distributed as source code. Its core utilities only depend on standard libraries to facilitate ease of compilation and use on multiple platforms. However, in addition to standard libraries, treeator may be compiled with the option to optimize model parameters under the maximum likelihood criterion using the NLopt library (Steven G. Johnson, The NLopt nonlinear-optimization package, http://ab-initio.mit.edu/nlopt); pairalign can be compiled in a parallelized version using pthreads, which may be useful to decrease runtime when doing pairwise alignment. As an addition to the four core programs rudisvg, a rudimentary svg viewer, that depends on the X11 library and a X11 server is included with phylommand (rudisvg).
The core programs of phylommand, in their basic version, have been successfully compiled and used on OS X 10.10 and Ubuntu Linux 16.04 (including Ubuntu 14.04 on Windows [anniversary edition of Windows 10]) using GNU g++ and make, and on Windows 10 using the same tools in MinGW. The svg viewer rudisvg has been successfully compiled and used on OS X 10.10 and Ubuntu 16.04 (Gnu/Linux; including Ubuntu on Windows). The behavior of the programs in phylommand are controlled by switches, and all programs have an extensive help documentation that can be accessed by the switch --help. Phylogenies can be given in either newick or nexus format. Pairalign reads DNA sequences in fasta format, and treeator reads character matrices/sequences in fasta, phylip, or nexus format (as outputted from for example AliView 1.19; Larsson, 2014) or work on distance matrices. The output formats for phylogenies are newick and nexus, but treebender can also output trees as standard vector graphics (svg). The svg output from treebender can be displayed by rudisvg. Treebender is faster at rooting trees than nw_reroot from Newick utilities (Table 1), but the difference is small compared to how much faster nw_reroot is for the same task compared with packages in interpreted languages such as BioPerl (Perl), APE (R), and ETE (python; Junier & Zdobnov, 2010).
In the examples, alignment_file.fst can be replaced by any fasta formatted alignment file, and tree_file.tree (tree_file.trees if including more than one tree) can be replaced by any newick or nexus formatted file. Treebender can easily be used to create a svg image that can be piped into a file:
treebender --output svg tree_file.tree > tree_file.svg
Treebender can also be used to create monophyletic operational taxonomic units in a tree based on branch lengths (c.f. virtual taxa sunsu Öpik et al., 2009):
treebender --cluster branch_length \ --cut_off 0.03 tree_file.tree
To view a neighbour joining tree representation of a set of DNA sequences it is possible to use:
pairalign -j -n -m -v alignment_file.fst \ treeator -n | treebender --output svg | rudisvg
The parsimony score and trees of all nearest neighbour interchange swaps from a topology can be given by:
treebender --nni all tree_file.tree \ -f alignment_file.fst -p
Pairalign can be used to get which taxa can be aligned confidently according to MAD scores:
pairalign --group alignment_groups \ -v alignment_file_with_taxon_string.fst
Contree can be used to draw the support values (e.g. bootstrap support) from a set of trees on a given topology:
contree -d tree_file.trees -a tree_file.tree
Contree can also be used to get supported conflicts between trees. The output can be either text or svg formatted as html (Figure 1):
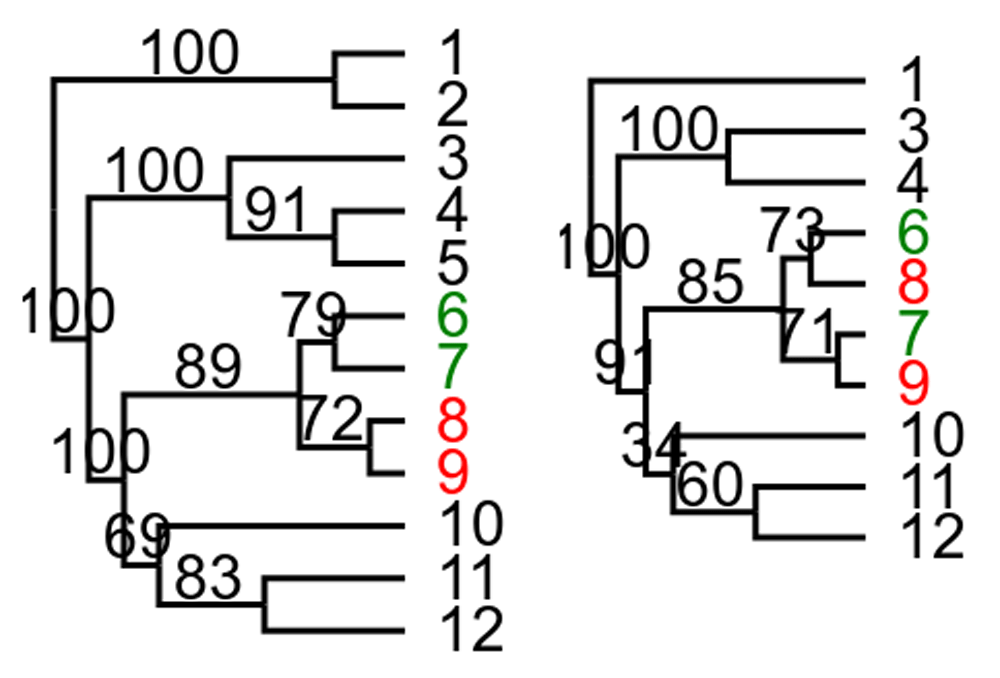
Part of output from contree -c 70 --html (re-formatted so trees are put next to each other and text removed). Tips in clade with more than 70 in support in tree to the left that is in conflict with tree to the right with more than 70 in support are colored green. The tips that cause the conflict in tree to the right are colored red.
contree -c 70 --html tree_file.trees
These and further examples, and an example of a bash script to do a search for the most parsimonious tree and a Perl script to find groups that are alignable according to MAD scores without a predefined taxonomy, are distributed with the source code.
Phylommand offers an efficient way of manipulating and analyzing phylogenetic trees without the overhead of a graphical interface or specialized command line interpreter. It can be used in both automated (through scripts) or manual work-flows. Since it is made to be compilable with minimum reliance of non standard libraries it is possible to use it on most operating systems including UNIX like systems as OS X and Linux, and Windows. This increases its utility for pipelines that will need to work on different platforms.
1. Latest source code available at: https://github.com/mr-y/phylommand
2. Archive source code as at the time of publication: http://doi.org/10.5281/zenodo.200397 (Ryberg, 2016)
3. License: GPL3
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)