Keywords
Variant calling, next-generation sequencing, NGS, exome, indel, validation
This article is included in the Data: Use and Reuse collection.
Variant calling, next-generation sequencing, NGS, exome, indel, validation
We recently released ICR142 Benchmarker, a tool that uses the ICR142 NGS validation series to generate standardised outputs and metrics to evaluate, optimise and benchmark variant calling algorithms. In the development of ICR142 Benchmarker we refined the ICR142 NGS validation series to maximise its utility, based on detailed re-review of all the data and user feedback.
The methods used in the data review are detailed in the amended paper and led to the exclusion of 26 sites such that the ICR142 NGS validation series now includes 704 sites: 416 sites with variants and 288 sites at which variants were called by an NGS analysis tool, but no variant is present in the corresponding Sanger sequence. This amended version of paper describes the updated dataset.
ICR142 Benchmarker is described in Ruark E, Holt E, Renwick A et al. ICR142 Benchmarker: evaluating, optimising and benchmarking variant calling using the ICR142 NGS validation series [version 1; referees: awaiting peer review]. Wellcome Open Res 2018, 3:108 (doi: 10.12688/ wellcomeopenres.14754.1)
To read any peer review reports and author responses for this article, follow the "read" links in the Open Peer Review table.
Next-generation sequencing (NGS) approaches have greatly enhanced our ability to detect genetic variation. Over the past decade NGS hardware, software, throughput, data quality and analytical tools have evolved dramatically. Thorough evaluation of each new laboratory and analytical development is challenging but necessary to fully understand how pipeline modification can impact results. To fully assess performance, NGS analysis tools should ideally be run on samples with pre-determined positive and negative sites assessed through orthogonal experimentation such as Sanger sequencing.
Over the past five years, we have generated extensive data on thousands of samples using different NGS instruments, sequencing chemistry, gene panels, exome captures and variant calling tools. Fortuitously, during this process we have generated orthogonal validation data using Sanger sequencing for a core set of 142 samples that were included in the majority of our experiments. We now formally use these samples, which we call the ICR142 NGS validation series, to evaluate NGS variant calling performance after any change to experimental or analytical protocols. This series has proved an extremely useful resource for our assessment of NGS analysis in both the research and clinical settings. We believe that it may also have utility for others, and hence are making it available here.
We used lymphocyte DNA from 142 unrelated individuals. All individuals were recruited to the BOCS study and have given informed consent for their DNA to be used for genetic research. The study is approved by the London Multicentre Research Ethics Committee (MREC/01/2/18).
Over the last five years we have generated data from the ICR142 validation series using different exome captures which we have analysed with multiple aligner/caller combinations1–6. To date we have generated Sanger sequence data for 704 sites amongst the 142 individuals. These sites include variants called by only one aligner and caller combination, increasing the representation of sites which can discriminate performance between methods.
To generate the Sanger sequence data, we performed PCR reactions using the Qiagen Multiplex PCR kit, and bidirectional sequencing of resulting amplicons using the BigDye terminator cycle sequencing kit and an ABI3730 automated sequencer (ABI PerkinElmer). All sequencing traces were analysed with both automated software (Mutation Surveyor version 3.10, SoftGenetics) and visual inspection.
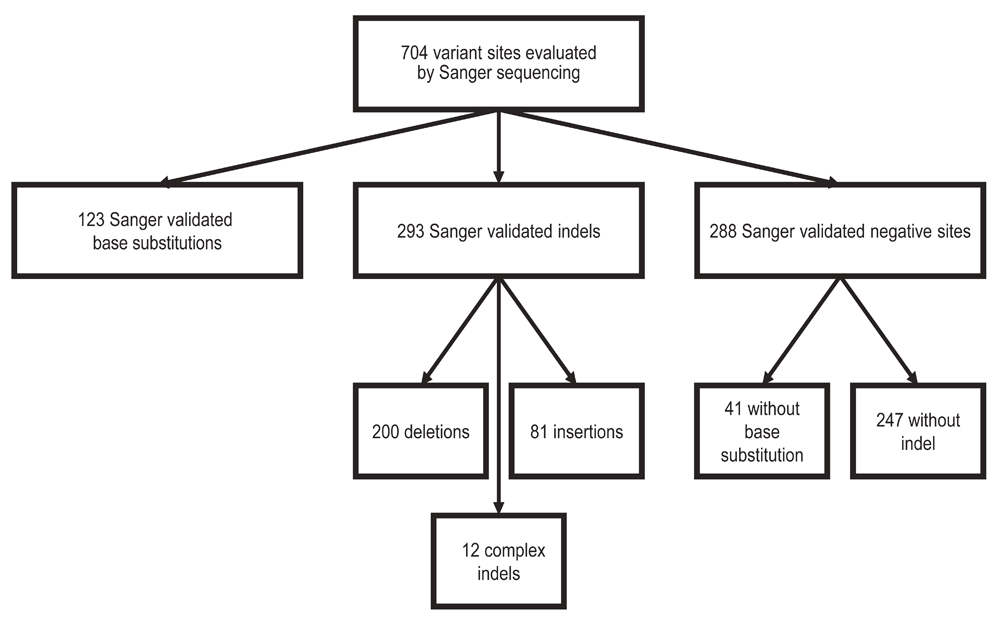
To determine if a variant was present we visually inspected each Sanger sequence with Chromas software v2.13. For each site we selected an ENST from release 65 as the reference sequence. We reviewed at least 100 base pairs of sequence flanking each variant site to allow for position/annotation errors. We considered a base substitution to be confirmed if the correct variant was called at the exact position and the variant base signal was accompanied by a corresponding reduction in the reference base signal. There were 123 confirmed base substitution variants. We considered an indel variant to be confirmed if an indel variant was present in the region of interest and the indel variant allele signal was present along the complete length of the region of interest. There were 293 confirmed indel variants.
We considered a site negative for a base substitution if the specific base substitution was not present, resulting in 41 negative base substitution sites. We considered a site negative for an indel if no indel, of any kind, was detected in the 200 base pair region of interest, resulting in 247 negative indel sites (Figure 1). We annotated confirmed variants with the HGVS-compliant CSN standard using CAVA (version 1.1.0) according to the transcripts designated in Supplementary table 17.
We have also generated high-quality exome sequencing data for the ICR142 NGS validation series. We prepared DNA libraries from 1.5 µg genomic DNA using the Illumina TruSeq sample preparation kit. DNA was fragmented using Covaris technology and the libraries were prepared without gel size selection. We performed target enrichment in pools of six libraries (500 ng each) using the Illumina TruSeq Exome Enrichment kit. The captured DNA libraries were PCR amplified using the supplied paired-end PCR primers. Sequencing was performed with an Illumina HiSeq2000 (SBS Kit v3, one pool per lane) generating 2×101 bp reads. CASAVA v1.8.1 (Illumina) was used to demultiplex and create FASTQ files per sample from the raw base call files.
All of the 704 sites had at least 15× coverage in the exome data, defined as at least 15 reads of good mapping quality (mapping score ≥20). Because these sites are well covered, we can readily assess the variant calling performance of any software tool by applying the pipeline to the exome sequencing data and comparing the variant calls with the Sanger sequencing dataset.
We have deposited the FASTQ files for all 142 individuals in the European Genome-phenome archive (EGA). The accession number is EGAS00001001332.
Researchers and authors that use the ICR142 NGS validation series should reference this paper and should include the following acknowledgement: "This study makes use of the ICR142 NGS validation series data generated by Professor Nazneen Rahman’s team at The Institute of Cancer Research, London”.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)