Keywords
proteomics, data-independent acquisition, mass spectrometry
proteomics, data-independent acquisition, mass spectrometry
For the last 15 years, liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based proteomics has provided broad detection and relative quantification—through chemical or metabolic labeling—of thousands of proteins across a variety of biological samples using a data-dependent acquisition (DDA) strategy1–4. In recent years, alternative LC-MS/MS targeted acquisition strategies, such as multiple-reaction monitoring (MRM)5–7 and parallel reaction monitoring (PRM)8, have provided precise and reproducible absolute quantification of up to hundreds of proteins. The ultimate goal of proteomics is the development of acquisition strategies that have both the breadth of DDA and the precision of MRM/PRM to provide reproducible identification and quantification of every protein in any biological sample. Although no single acquisition strategy can yet achieve this goal, recent advances in hardware and software show that a recently resurfaced strategy9, data-independent acquisition (DIA), may provide a viable path to this goal10. Below is a discussion of DDA, MRM/PRM’s shortcomings, DIA’s circumvention of these shortcomings, current software to analyze DIA spectra, and efforts to further improve DIA analysis.
All LC-MS/MS methods discussed in this article are bottom-up proteomics: Proteins are enzymatically digested into peptides which then are separated using high-performance liquid chromatography (HPLC), ionized, isolated, fragmented, and detected in the mass spectrometer as they elute from the HPLC. HPLC delivers peptides into the mass spectrometer for a period of time (tens of minutes to a few hours, depending on the application), separating the peptides according to their physicochemical characteristics4,11, thus increasing sample coverage. In LC-MS/MS methods, three events occur in the mass spectrometer: (a) ionization: peptides elute into the mass spectrometer from the HPLC and are ionized; (b) MS1 scan: the abundance and mass-to-charge ratios (m/z) of all ions eluting at a given time are measured; and (c) MS2 scan: some or all detected ions are fragmented, and the abundances and m/z’s of the fragments are measured and recorded. Different LC-MS/MS methods vary in how ions are selected and measured in the MS2 scan. Figure 1 shows a cartoon schematic of how peptides are isolated, fragmented, and analyzed by a mass spectrometer working on DDA, MRM, PRM, or DIA modes.
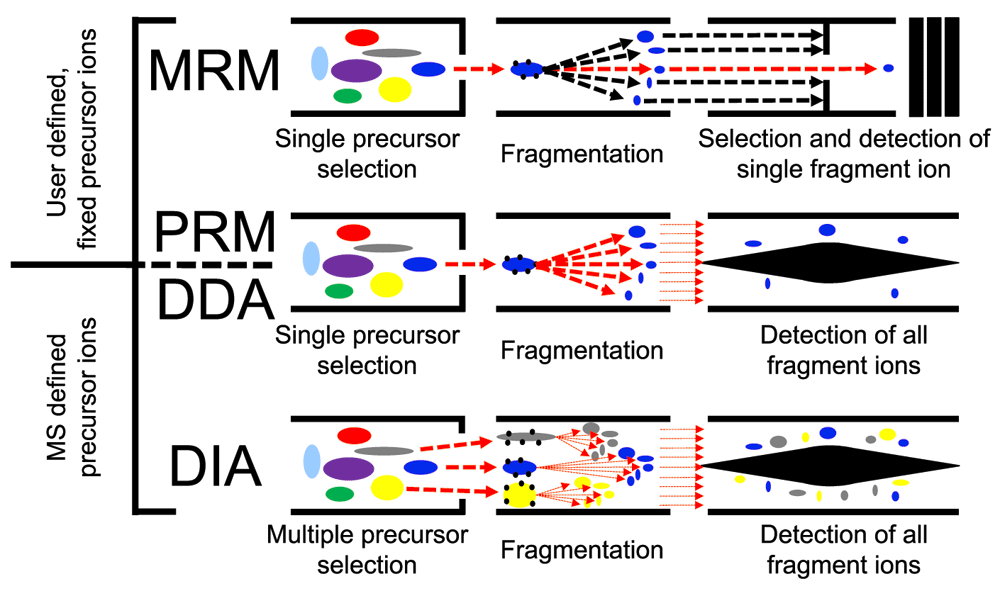
In DDA, MRM, and PRM, single precursor ions are isolated, fragmented, and analyzed in an MS2 scan by the mass spectrometer. In DDA mode, the precursor ions are chosen by the instrument on the basis of abundance. In MRM and PRM, the precursor ions to be analyzed are fixed by the user. DIA is different form the methods above in that all precursor ions within a selected mass range are isolated, fragmented, and analyzed in a single MS2 scan. MS1, scan in which the peptide ions entering the mass spectrometer at a given time are identified; MS2, scan in which the fragments of all (or some) of the peptides that are in the mass spectrometer at a given time are identified.
In DDA, a subset of the most abundant ions reaching the mass spectrometer detector during an MS1 scan are individually isolated and fragmented in sequential MS2 scans (Figure 1 and Figure 2A), and each MS2 scan (Figure 2C) can be analyzed with a database search algorithm1,4. Currently, most instruments can perform a DDA cycle with one MS1 scan and 10 MS2 scans within 2 seconds. DDA typically yields thousands of protein identifications. Unfortunately, irreproducibility and imprecision are fundamental to DDA’s design; if too many peptide species co-elute and appear in a single MS1 scan, then DDA stochastically samples only the most abundant peptides and misses the rest. This approach diminishes reproducibility and prevents the measurement of low-abundance peptides9. Additionally, to survey as many peptides as possible, DDA deliberately samples each peptide species only once or twice, preventing precise absolute quantification that requires multiple measurements per peptide. DDA analysis has been used for a variety of studies, including the characterization of epidermal growth factor receptor (EGFR) signaling networks12,13, the characterization of the proteome on different mouse organs14,15, identification of protein interaction partners16–18, and description of the role of viral infections in modulating host proteomes18–21, which had been thoroughly covered in a special issue of the journal Proteomics22. In spite of its flaws, DDA’s flexibility, breadth of detection, and the simplicity of its setup and analysis, make DDA the preferred LC-MS/MS method among the wider scientific community. Additionally, DDA allows relative quantification of peptides between selected samples through a variety of chemical labeling schemes—e.g., isotopic (stable isotope labeling by amino acids in cell culture, or SILAC)23 or isobaric (isobaric tags for relative and absolute quantitation, or iTRAQ)24 labeling.
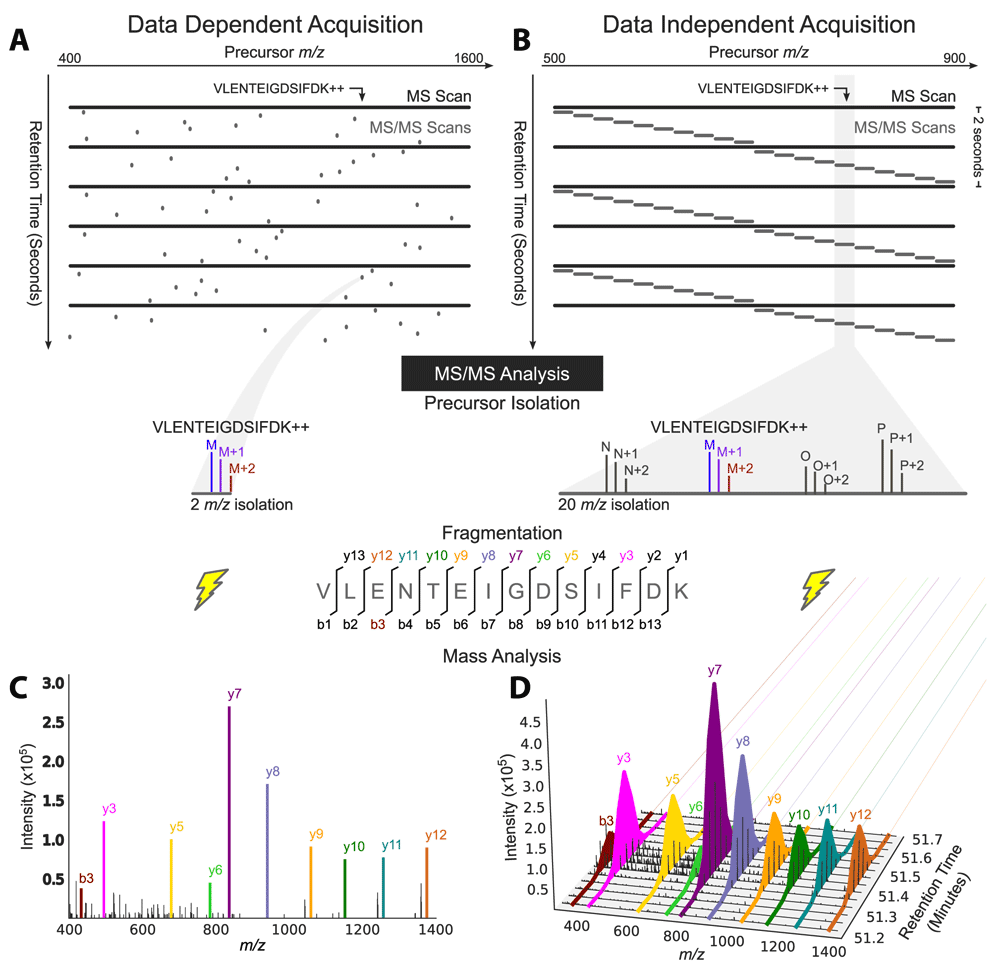
(A) Data-dependent acquisition (DDA) acquires MS/MS scans with narrow isolation windows centered on peptide precursors detected in an MS scan over a wide range of masses: 400 to 1,600 mass-to-charge ratio (m/z) here. (B) Data-independent acquisition (DIA) acquires MS/MS scans with wide isolation windows that do not target any particular peptide precursor. Instead, the scans are arranged side-by-side to collectively cover a desired precursor m/z range (500 to 900 m/z here) comprehensively, and several precursors are fragmented together in a single MS2 event (four here: identified peptide M and peptides N, O, and P). (C) Fragment ion information for the peptide precursor VLENTEIGDSIFDK++ is present in a single MS/MS spectrum in a DDA analysis, (D) but it can be extracted over time from DIA data and used for quantification owing to the repetitive MS/MS sampling cycle of DIA. Adapted with permission from Egertson et al.46.
Alternatively, the targeted methods MRM and PRM avoid the imprecision and irreproducibility of DDA by focusing MS2 scans on only a small set of predetermined and previously identified peptides. Instead of selecting the top n precursors in an MS1 scan for further fragmentation in MS2 scans, these methods select only precursors and fragments with the m/z and elution time that match a pre-specified peptide of interest. The knowledge of a peptide’s elution time, MS1 m/z value, and robustly detectable fragments is determined prior to the MRM/PRM experiments by previous DDA identification or MS/MS measurement of its synthetic version in a simplified background or both. In MRM, for each MS1 scan, a subset of fragment ions is measured in the subsequent MS2 scan25, whereas in PRM, all of the fragment ions are measured8. In both, the same precursors are selected and fragmented multiple times to acquire more precise quantification of fewer peptides, compared with DDA. MRM was developed first and has been shown to robustly quantify tens of blood plasma biomarkers of low abundance across laboratories toward clinical use6 and tens of low-abundance transcription factors and kinases in human cells7. Currently, the Clinical Proteomics Tumor Analysis Consortium (CPTAC)26 database hosts a collection of 679 MRM assays for human proteins. PRM, which succeeded MRM, outperforms it in terms of throughput and absolute quantification thanks to the use of high-resolution spectrometers capable of parallel fragment analysis8,27. PRM is still evolving; however, it has already been used successfully to address difficult proteomics problems in human health, such as the role of low-abundance polyubiquitin chains28 in Parkinson’s disease29, and in plant biology to monitor the degradation of low-abundance peptides in Arabidopsis thaliana30.
DIA is like MRM/PRM in that it repeatedly samples the same peptides for more precise quantification, but it differs from them and DDA by dispensing with the isolation of individual peptide species and instead repeatedly selecting mixtures of peptide species within large, pre-specified mass ranges (Figure 1 and Figure 2B) for MS2 scans. DIA is therefore guaranteed to sample all peptides within the selected mass ranges, allowing for the identification of all sufficiently abundant peptides within them if the resulting spectra are properly interpreted10.
Proper interpretation of DIA data is currently problematic because the complex MS2 scans contain mixtures of peptides and therefore are more difficult to analyze. Fortunately, recent developments in bioinformatics software have adequately overcome this DIA issue, so that DIA now closely matches DDA in the number of peptide identifications while still allowing precise quantification of most of them. Quantification relies on comparing DIA spectra to sets of annotated and refined peptide-MS2 spectrum matches from DDA experiments (or the same DIA experiment in DIA-Umpire’s algorithm) called spectral libraries that show accurate, empirically determined fragmentation patterns for each peptide in the library. However, DIA is currently unable to match the precision of MRM or PRM in measuring very low-abundance peptides, likely because their signals are dwarfed by those from abundant co-eluting peptides. A brief comparison of DDA, DIA, and MRM/PRM with respect to precision of quantification, breadth of identification, ease of setup and analysis, and reproducibility, is shown in Table 1.
| Method | Instrument setup | Ease of data analysis | Precision of peptide quantification | Reproducibility of peptide identification | Breadth of peptide identification |
|---|---|---|---|---|---|
| DDA | Easiest Requires user selection of precursor m/z range and frequency of precursor scans. Is the default mode of use on most commercial instruments. | Easiest Many convenient and comprehensive pipelines for the analysis of DDA spectra have been developed over more than 20 years48,51,52. | Low/Moderate/High Spectral counts (low), isobaric-tag labels (moderate), or SILAC (high) can be used for relative quantification of protein abundance across samples. Hard to use for absolute quantification. | Lowest Run-to-run peptide identification overlap for a given sample is around 60%53. | Highest Samples and identifies a single time as many peptides as can be individually isolated. |
| PRM/MRM | Hardest Requires prior identification of peptides and, in MRM, selection of reproducible fragments that do not exhibit interference54. | Moderate A few pipelines have been developed over the past few years but require some manual curation to identify and quantify fragment chromatograms54. | Highest Provides good relative peptide quantification and can be coupled with heavy labeled reference peptide for absolute quantification. Most sensitive method because of high signal-to-noise ratio54. | Highest Run-to-run peptide identification overlap for a given sample is more than 85%54. | Low Repeatedly samples and identifies a small set of pre-specified peptides54. |
| DIA | Easy Requires user selection of precursor m/z windows for MS1 and MS2 scans. | Hardest Requires multiple steps from multiple experiments to compile spectral libraries, with more parameters to choose in recently developed, not-yet- established pipelines. | Moderate/High Similar to PRM/MRM but more vulnerable to variation caused by interference from other peptides32–37,39–45,47,50–54. | High Similar to PRM/MRM but more vulnerable to variation caused by interference from other peptides32–37,39–45,47,50–54. | High Repeatedly samples every peptide within pre-specified m/z windows and identifies those whose signals can be successfully deconvolved54. |
DDA, data-dependent acquisition; DIA, data-independent acquisition; MRM, multiple reaction monitoring; MS1, scan in which the peptide ions entering the mass spectrometer at a given time are identified; MS2, scan in which the fragments of all (or some) of the peptides that are in the mass spectrometer at a given time are identified; m/z, mass-to-charge ratio; PRM, parallel reaction monitoring; SILAC, stable isotope labeling by amino acids in cell culture.
The complexity of MS2 spectra greatly impacts the sensitivity of the downstream analyses and therefore must be considered when planning a DIA experiment. Two main variables determine the complexity and interpretability of the spectra: the number of proteins present in the sample and the precursor m/z window widths from which ions are isolated and fragmented. The effect of the number of proteins present is shown in Figure 3, which describes the results of a study in which 345 synthetic peptides were spiked into water, yeast lysate, or human lysate backgrounds in concentrations varying from 30 to 0.058 fmol/µL and then analyzed via relatively wide precursor isolation windows of 25 m/z31 (see Supplementary material). The sensitivity of peptide detection decreases with the protein complexity of the organism, where the yeast lysate is more complex than the water background and the human lysate is more complex than the yeast lysate, with an average of 32,993 unique human, trypsin-digested peptide ions possibly falling within each 25-m/z window compared with the 7,287 yeast peptide ions. Sensitivity and precision may be increased by shortening the width of the precursor isolation window, thereby decreasing the number of peptides represented in each MS2 spectrum. However, this decreases coverage across the total precursor m/z range and therefore the number of peptides to which the analysis may be sensitive. Therefore, experimentalists must balance the effects of sample complexity, isolation window width, and desired coverage on sensitivity.
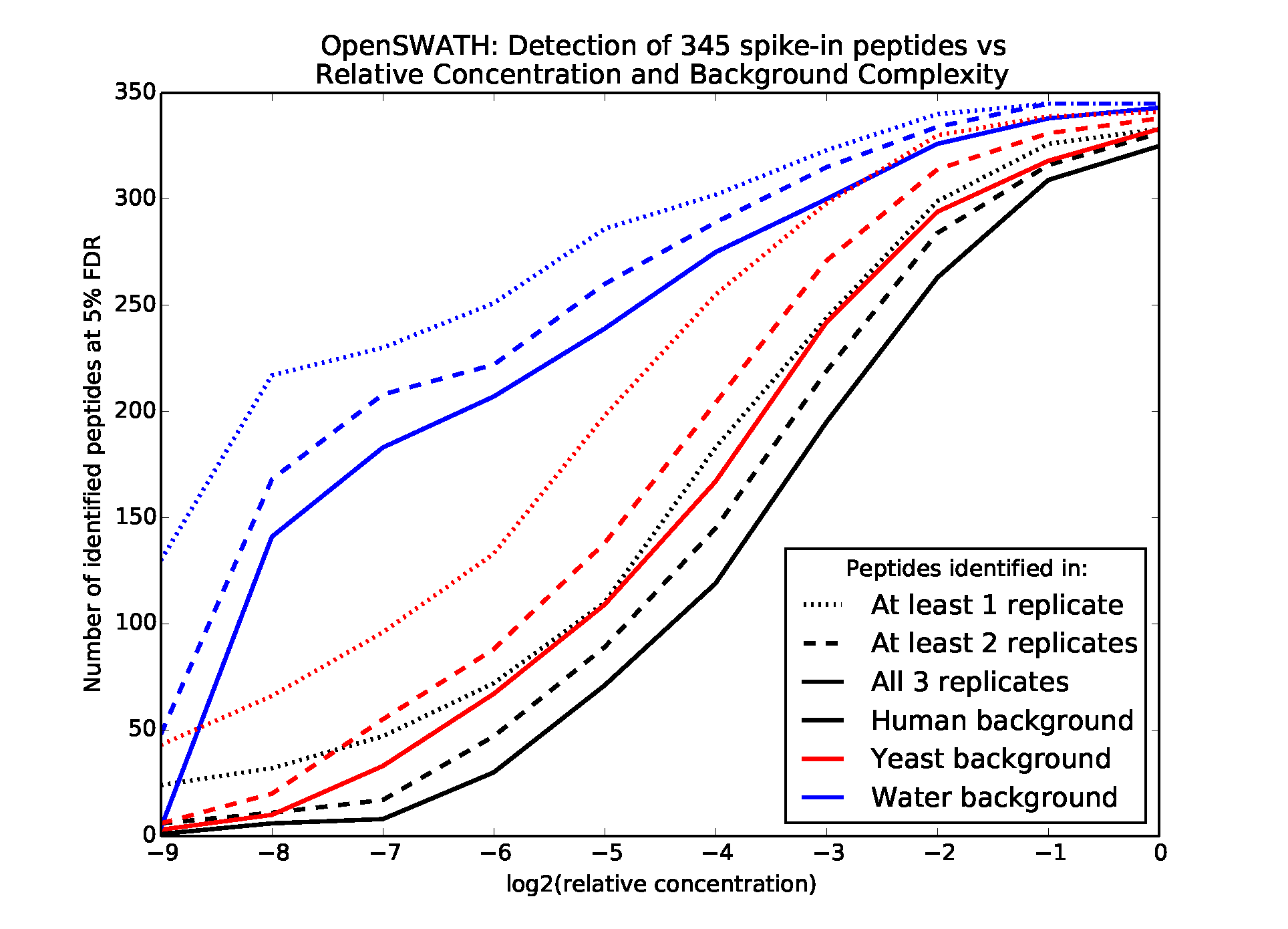
Varying concentrations of 345 synthetic peptides were spiked into three sample backgrounds, subjected to data-independent acquisition (DIA), and analyzed by OpenSWATH. Lines show the number of spike in peptides identified at a 5% false discovery rate (FDR) in the different samples.
The main reason for the decrease in sensitivity is the increased likelihood of fragment ion interference in complex spectra, which occurs when multiple co-eluting peptides share a fragment ion peak. Interference undermines the elution profile correlation between a peptide’s fragments and the fragments’ correlation to spectral libraries on which many DIA analysis methods rely. In an analysis of synthetic peptides spiked into human urine, SWATHProphet32, a software tool further described below, estimated that at least 24% of confidently identified peptides showed evidence of fragment ion interference that increased the variance of their quantification. This percentage is likely much higher in unidentified peptides and this is the likely cause of the peptides’ invisibility to the software. Therefore, further progress in the interpretation of DIA spectra should circumvent the problem of interference, which SWATHProphet32 has begun to do by identifying and disregarding fragments affected by interference.
In all DIA methods, each MS2 scan contains fragments from every peptide within one or more pre-specified precursor m/z windows. Each window is repeatedly sampled so that each peptide is fragmented multiple times. The earliest and most common DIA method is a sequential sampling strategy9,33 (Figure 2B), in which an m/z range covering most peptides of interest is split into a sequence of non-overlapping windows, usually of equal but sometimes of variable size depending on the m/z distribution of the peptides of interest. For each window in the sequence, all of the precursors falling into that window are fragmented together and measured in an MS2 scan. The machine repeats the sequence throughout the full HPLC elution gradient. The time needed to complete the traversal of the sequence is on the order of a few seconds, such that every peptide can be sampled at least a few times during its elution.
An alternative DIA method, MSX34, incorporates multiplexing and an element of randomness to the sequential sequencing to increase precision in associating a precursor ion to its fragments. However, this method is compatible only with selected mass spectrometers in which software controllers have been modified to accept random sampling of mass range windows. The precursor m/z windows are smaller and more numerous than in the sequential method, but each MS2 scan is multiplexed. Each scan contains fragmented precursors from multiple, randomly chosen non-contiguous windows, such that the precursors span the same total m/z length in each MS2 scan. Post-processing of the MS2 spectra solves a system of linear equations to infer from which smaller precursor window each fragment ion peak originated. The resulting de-multiplexed MS2 scans allow a modest increase in peptide identifications compared with scans produced from non-random, contiguous windows.
Although traditional algorithms for identifying peptides from DDA spectra can be applied to DIA spectra analysis, these algorithms are not appropriate for DIA for two reasons: they incorrectly assume that each MS2 scan contains fragments from just one peptide, and they ignore the dynamic pattern of elution profiles in DIA spectra. Consequently, three main classes of computational algorithms have emerged to specifically analyze DIA data that accommodate the complexity and time variation in DIA spectra. The first two classes are for untargeted, discovery-based identification, and the third is for precise quantification of previously identified peptides from spectral libraries. The main challenge of the algorithms below is controlling the false discovery rate among the identified peptides while identifying all (or most) real peptides in the sample of interest.
These methods use a pre-processing step that deconvolves DIA MS2 scans into multiple pseudo-spectra, each containing the fragments of only a single peptide species in the mixture. The intensities of different fragments of the same peptide species should correlate over elution time, and the pre-processing step uses this correlation to assign fragment ions from MS2 scans to their intact peptide species in MS1 scans. These pseudo-spectra then can be searched by using a traditional DDA database search method.
DIA-Umpire35 and DeMux36 are two strategies that take this approach. They differ in the specific algorithms used to group ions and compile them into deconvolved spectra. DIA-Umpire tends to work better because it considers isotope peak distributions in MS1 scans to narrow down candidate peptides, finding up to 89% of the peptides identified by analogous DDA experiments. DIA-Umpire also includes additional methods that generate new reference/library spectra, incorporates prior library spectra, and uses them for further steps in protein identification and quantification, achieving on average a 0.931 R2 correlation in the quantifications of peptides between replicates. This strategy works in a fashion analogous to DDA experiments in that it identifies many peptides, but also the strategy provides precise DIA quantification of the identified peptides35.
These strategies are inspired by traditional database searching and do not focus on using dynamic patterns to identify peptides. Instead, they include other heuristics to adapt searching to a DIA context. They score each peptide against each observed spectrum by computing the dot-product of the peptide’s theoretical spectrum against each observed spectrum, much like traditional DDA search algorithms. However, these methods introduce additional heuristic filtering steps, such as considering only observed spectra that match a threshold number of peaks in a theoretical spectrum. The first example of this strategy was FT-ARM37. However, a more recent method, Pecan38, contains additional heuristics as well as a discriminative model to combine them, so it performs better than FT-ARM. Pecan is available through the Skyline34–37,39 graphical user interface, which provides convenient visualization, annotation, and analysis of mass spectra acquired by DDA, DIA, and MRM/PRM.
Strategies in this category are adapted from methods used to analyze PRM/MRM spectra, because both DIA and MRM/PRM repeatedly sample the same peptides to obtain sequences of fragment ion intensities over elution time (fragment ion chromatograms; Figure 2D). These methods take library spectra as input (compiled from prior DDA peptide identifications or prior DIA-Umpire identifications) and extract fragment ion chromatograms at the peaks in the library spectra. Potential elution peaks of each peptide from these fragment ion chromatograms are evaluated on the basis of many criteria, including how well the fragment ions correlate over elution time and how well their relative intensities match their corresponding library spectrum. Elution peaks are evaluated by using a discriminative model that combines these criteria to distinguish real peptide signals from decoy peptide signals. These methods also quantify proteins by quantifying the fragment ion chromatograms of their peptides.
OpenSWATH31, SWATHProphet32, Spectronaut40, and a module in DIA-Umpire35 all implement this strategy. Skyline39 provides elution peak quantification but not statistical validation. OpenSWATH31 has been shown to achieve coefficients of variation of between 0% and 20% on 345 spike-in peptides across 256-fold concentration differences in a yeast lysate. In a series of human lysate runs, Spectronaut40 achieved a 98% peptide identification reproducibility rate from run to run on DIA data compared with 49% on DDA data on 26,738 peptides covering 3,690 proteins.
The class of search methods to be used depends on the specific context of the experiment. If no library spectra are available, then Pecan or DIA-Umpire must be used. No published direct comparison yet exists between the two, so the choice depends on how they fit into your bioinformatics pipeline. DIA-Umpire includes its own pipeline for library spectrum generation and automated quantification. Pecan is incorporated into Skyline39 such that visualization, annotation, and semi-automated quantification are convenient. If library spectra are available, then a method with chromatogram scoring should be used. Again, no direct comparison is available, but the different chromatogram scoring algorithms are similar enough such that one can use the tool that best fits into one’s pipeline.
Here, we describe the use of algorithm 3, chromatogram scoring with spectral libraries, on two large quantitative studies whose insights critically depend on the breadth, reproducibility, and quantitative precision of DIA. The first study characterizes plasma proteome variation between monozygotic and dizygotic twins41 and elucidates biomarker variation over time. Its inclusion of both types of twins allows the quantification of variation caused by genetics, the environment, and time. The study sampled, at two time points, blood from each person in 22 pairs of adult fraternal twins and 36 pairs of adult identical twins, resulting in 232 sets of DIA experiments. The spectral library used to identify and quantify peptides consists of 43,000 peptides that represent 1,667 unique proteins, compiled from a pre-existing library42 and supplementary DDA data. OpenSWATH analysis was able to identify 1,904 of these peptides (342 proteins) in all 232 samples and quantify 76% of these with coefficients of variation of less than 25%. Notably, 42 of the identified proteins are approved by the US Food and Drug Administration for clinical assays. The breadth and precision of quantification vastly surpassed prior attempts using antibody arrays43 and could be achieved only by using DIA rather than PRM or DDA.
A thorough survey of the successfully identified proteins characterized the main causes of their variability: proteins involved in blood coagulation, inflammation, and high-density lipoproteins that regulate cholesterol levels are controlled more by genetics than environmental influences. These results corroborate findings from several prior studies. Moreover, the survey discovered previously uncharacterized dependencies of eight of the clinically relevant proteins on age, which could confound their clinical interpretation. For example, plasma level of soluble CD14 is used as an independent predictor for HIV infection, and 14-3-3 protein zeta/delta is used as a prognostic for lung and breast cancers; both of these proteins naturally vary over time41.
In a second study, DIA and OpenSWATH successfully mapped the interactomes of four well-characterized human proteins via affinity purification of Flag-tagged proteins and discovered how the interactions of two of them with the chaperone protein HSP90 differ in response to melanoma-associated mutations44. In particular, DIA analysis of CDK4 affinity-purified samples out-identified and out-quantified analogous DDA experiments: 5,089 peptides were identified in all three DIA replicates, whereas 2,741 were identified in all three DDA replicates. Of peptides identified by both DIA and DDA, DIA quantified 82.1% with a coefficient of variation of less than 20% compared with the 74.5% achieved by DDA. Samples affinity-purified for melanoma-associated mutant versions of CDK4 showed increases in HSP90 abundance, suggesting that these mutants associate more or form a stronger association than wild-type CDK4 with HSP90.
Although DIA has been gradually accepted by the proteomics community, improvements in hardware and software tools are still required to facilitate its use by the larger scientific community. From a hardware standpoint, parallel improvements in duty-cycle speed, sensitivity, and peak resolution, especially at the MS2 level, will be critical for the improvement of DIA. Faster duty-cycle speed could allow increases in the mass regions analyzed by a DIA experiment in a single run. Currently, DIA experiments typically range from 100 to 400 m/z units (500 to 900 m/z region), whereas DDA usually covers 1,600 m/z units (400 to 2000 m/z region). Moreover, increases in duty cycle would allow a reduction of the size of the fragmentation windows on DIA methods from 15 to 25 m/z units on average to 2 to 5 m/z units without needing to reduce the mass range to be analyzed in a single run. Much like MRM methods, isolating small regions for fragmentation would result in increased signal-to-noise ratios at the MS2 level, which combined with increased sensitivity would allow the detection of fragment ions formerly lost in the noise. Moreover, the use of smaller fragmentation windows combined with increases in instrument resolution, currently provided by high-end instruments, would result in improved deconvolution of mixed spectra because the mixtures are simpler (fewer peptides fragmented in each MS2 event) and fragments that are close in mass are easier to differentiate (higher resolution). No mass spectrometers yet combine the resolving power and speed needed to cover the same mass range that DDA does using small windows for DIA analysis; however, mass spectrometry speed, sensitivity, and resolution have greatly improved over the last 5 years, and we are yet to reach the physical limits of hardware improvements45. If the rate of instrumentation advances continues, then within the next 5 years we should be able to cover the same m/z range that DDA covers using small, overlapping DIA windows. Thus, the main challenge is how these large DIA datasets will be deconvoluted and analyzed.
To further improve the deconvolution of complex DIA spectra and increase their identification and quantification efficiency, researchers are developing more sophisticated sampling and computational algorithms to analyze biological samples using DIA. Though yet unpublished, several such methods were presented at the American Society of Mass Spectrometry conference. Three of the most promising analysis methods are outlined below.
The first method draws a parallel between the analysis of DIA spectra and the field of compressed sensing to precisely infer the precursor masses of the detected fragment ions46. It uses the random, multiplexed sampling of MSX34 but improves the deconvolution of multiplexed spectra by adding further constraints to the system of linear equations posed by deconvolution, inspired by the field of compressed sensing. Compressed sensing takes advantage of the fact that if a signal is sparse, then one need not measure all of the signal to accurately reconstruct it47. DIA data are sparse; at any time during the chromatography, only a small fraction of possible precursor peaks is observed. To precisely match precursor ions to their fragments, it is unnecessary to dedicate fragment scans to all small individual precursor windows if most windows are devoid of peptides. Indeed, DIA methods use wide, contiguous precursor isolation windows of width up to 25 m/z. However, compressed sensing states that repeated sampling from these predetermined windows are suboptimal to match fragment ions to their precursors. Instead, if one randomly combines smaller, not necessarily adjacent windows into large composite windows that overall span 25 m/z, then one can match fragments to precursors within these smaller, more precise windows. The published MSX34 method already leverages this fact but does not use the theoretical techniques developed in compressed sensing to get the most accurate deconvolution.
The second method attempts to improve identifications by using linear regression to jointly identify the whole set of present peptides simultaneously rather than one at a time48. Unlike current methods, the regression can deconvolve fragment ion interference in a principled way, which occurs when multiple precursor ions contribute to intensity at the same fragment ion peak, such that its chromatographic profile no longer matches the chromatographic profiles of the other fragment ions. Estimates have shown that approximately 24% of peptides may exhibit interference in a human urine sample32. This phenomenon prevents current methods from attributing the fragment ion peak to any single precursor ion, but the regression approach can in principle properly attribute the peak to all appropriate precursor ions simultaneously.
The third method extends traditional DDA search methods with peak filtering to take into account the correlation of fragment ion peaks from the same peptide49. When scoring matches between candidate peptides and observed MS2 scans, some shared peaks can be explained by multiple candidate peptides and some unique peaks can be explained by just one. The method restricts each shared peak to contribute to the score of only the single peptide whose unique peaks correlate best over time with the shared peak. This restriction prevents the spurious high scores of falsely discovered peptides that depend entirely on peaks originating from other peptides. These techniques to better associate fragment ions to their precursor ions are in developmental stages and, if successful, will broaden the usefulness of any DIA dataset.
Because DIA combines the breadth of protein identification provided by DDA and approaches the sensitivity and precision of MRM/PRM, it will be the best choice for discovery bottom-up proteomics and large-scale quantification in the near future. Sampling schemes and analysis methods already allow flexibility to adapt DIA to most biological problems and researcher needs. However, because DIA is still a work in progress, DDA data are still easier to acquire and analyze for most researchers and DDA is the method of choice for most biology and proteomics laboratories. Finally, using DIA implies trading off some precision and sensitivity for breadth when compared with targeted methods. Thus, if quantification of specific lowly abundant peptides is required, then MRM and PRM targeting the ions of interest are still the better option.
DDA, data-dependent acquisition; DIA, data-independent acquisition; HPLC, high performance liquid chromatography; LC-MS/MS, liquid chromatography-tandem mass spectrometry; MRM, multiple reaction monitoring; MS1, scan in which the peptide ions entering the mass spectrometer at a given time are identified; MS2, scan in which the fragments of all (or some) of the peptides that are in the mass spectrometer at a given time are identified; m/z, mass-to-charge ratio; PRM, parallel reaction monitoring.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)