Keywords
Rho-ROCK, actomyosin, metastasis, Rho
Rho-ROCK, actomyosin, metastasis, Rho
Metastatic disease is still largely incurable because of its systemic distribution and resistance to current therapies, and it is the cause of more than 90% of cancer-related deaths1,2. In spite of its clinical importance, the underlying cellular and molecular mechanisms of cancer metastasis are only partially understood3. Thus, improved knowledge of how cancer cells acquire metastatic traits is necessary to unravel novel drug targets and prognostic markers of distant relapse.
Metastasis is a complex multi-stage process by which cancer cells disseminate from primary tumors, survive in distant sites and eventually grow as secondary tumors3. The main events of the metastatic cascade involve loss of cell-cell contacts, cancer cell migration, local invasion of the surrounding extracellular matrix (ECM), interactions with stroma, intravasation and transit into blood or lymphatic vessels, arrest at secondary sites, extravasation, survival and colonization of distant sites4. Genetic alterations and deregulation of critical oncogenic signaling pathways affecting survival, proliferation, apoptosis, and cell motility, regulate many of these complex metastatic events3,5. In addition, the interaction with the tumor microenvironment such as ECM, growth-supportive stromal cells, inflammatory cells and endothelial cells strongly impacts the metastatic capabilities of cancer cells6,7.
Many signaling pathways have been reported to have an impact on metastasis and have been the focus of excellent reviews8–15. In the present review, we will focus on Rho-ROCK signaling and actomyosin contractility, key regulators of several main steps in metastasis. Rho-ROCK, through its actions on cytoskeletal dynamics and through regulation of critical signaling pathways, controls several cellular processes important for metastasis such as cell migration, local invasion, survival at the secondary site, and tumor outgrowth16–18.
The Rho family of small GTPases plays crucial roles in the regulation of the actin cytoskeleton, cell polarity, cell migration, cell proliferation, invasion, and metastasis19. Rho GTPases act as molecular switches cycling between a guanosine triphosphate (GTP)-bound active state and guanosine diphosphate (GDP)-bound inactive state to translate extracellular signals into different cellular responses19. Their activity is controlled by guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs)18. The best studied and most conserved Rho family members across eukaryotic species are Ras-related C3 botulinum toxin substrate 1 (Rac1), cell division control protein 42 homolog (Cdc42), and Ras homolog gene family member A (RhoA)18. Rac1 stimulates lamellipodia formation20, whereas RhoA regulates the formation of stress fibers or favors amoeboid migration depending on the cellular context and the properties of the matrix. RhoA bound to GTP leads to activation of its effectors Rho-associated protein kinases (ROCK1 and ROCK2)21–23. ROCK1/2 serine/threonine kinases promote actomyosin contractile force generation by decreasing myosin phosphatase activity and thereby increasing phosphorylation of myosin light chain 2 (MLC2)24. On the other hand, Cdc42 induces filopodia formation25, but Cdc42 signaling can also generate actomyosin contraction through p21 protein (Cdc42/Rac)-activated kinase 2 (PAK2) and myotonic dystrophy kinase-related Cdc42-binding kinase (MRCK) kinases26,27. Deregulation of the Rho-ROCK signaling pathway has been found in a variety of cancer types and in several cases correlates with disease progression28–30 (Table 1). Furthermore, inhibition of ROCK signaling could suppress migration and invasion in vitro and impair the metastatic process in vivo, suggesting that ROCK inhibitors might be potential anti-metastatic agents30–32.
Shown are examples in the literature of where different stages of the metastatic cascade have been shown to be influenced by Rho-ROCK and/or actomyosin contractility signalling. (SCC= Squamous cell carcinoma)
| Cancer type | Step of metastatic dissemination | Reference |
|---|---|---|
| Breast, colon | Local invasion and migration | 40,46,52,68 |
| Breast | Intravasation | 40,66,71 |
| Breast | Survival in circulation, adhesion to vessels, early lung colonization | 92 |
| Oesophageal | Invasion and survival in circulation | 104 |
| Lung | Transendothelial migration | 81 |
| Prostate | Transendothelial migration | 75 |
| SCC | Fibroblast mediated invasion and migration | 42 |
| Melanoma | Local invasion and migration | 23,32,45,46,49,105 |
| Melanoma | Intravasation, extravasation, survival in circulation, adhesion to vessels, early lung colonization | 23,32,48,54,58,92,95–97,105 |
The ability of cancer cells to migrate into and invade surrounding tissue is a critical step in the metastatic cascade, which requires increased cell motility driven by altered cytoskeletal organization and contacts with the ECM and the stroma33. Cancer cells can move either collectively or as individual cells34,35. The majority of tumors originate from epithelial tissues, and epithelial cancer cells that leave the primary tumor undergo a complex program called epithelial-mesenchymal transition (EMT). Incomplete or partial EMT allows collective migration in which cells can maintain cell-cell adhesions and migrate collectively in a coordinated manner as strands, sheets, or cell clusters. On the other hand, complete EMT is associated with the loss of cell-cell adhesions in favor of cell-ECM interactions and the concomitant acquisition of individual migratory characteristics36,37. After undergoing EMT, individual cancer cells can engage into elongated mesenchymal or rounded amoeboid modes of movement, distinguished by their different usage of signaling pathways. Mixed mesenchymal and amoeboid phenotypes have also been identified38,39. Individual cell migration seems to be required for blood-borne metastasis40.
Actomyosin contractility driven by Rho or ROCK signaling is key in controlling tumor dissemination, as all forms of cell migration require a certain degree of actomyosin force34,41. During collective cell migration, actomyosin contractility is high around the edges of groups of invading cancer cells, which generates pulling forces between the substrate and the follower cells, together with a prominent actomyosin ring at lateral regions of the groups to maintain coupling between cells and collective forward movement42,43. On the other hand, in individual migration, the contractile cortex is crucially important for amoeboid to intermediate forms of movement, and some degree of contractility is also required to retract protrusions in mesenchymal migration39,44–46. The mesenchymal mode of movement is characterized by an elongated, spindle-like shape, high levels of adhesion, and Rac-dependent adhesive actin-rich protrusions23,46,47. On the other side of the spectrum, in amoeboid migration, cancer cells adopt a rounded or irregular morphology with blebs as functional protrusions. Amoeboid motility is promoted by high levels of RhoA/Ras homolog gene family member C (RhoC) or ROCK-driven actomyosin contractility and requires lower levels of adhesion that allow higher speeds of movement46–50.
Cancer cell migration is a dynamic process, and individual cancer cells can switch between modes of movement to adapt to the changing microenvironment and facilitate tumor dissemination. Different cues will favor either a mesenchymal-amoeboid transition (MAT) or an amoeboid-mesenchymal transition (AMT)23,45,49,51,52. Their core regulatory network is the mutually inhibitory circuit between Rac1 and Rho GTPase signaling in migrating cells (Figure 1). Higher Rac1 activity promotes cell elongation and permits long actin-rich protrusions characteristic of mesenchymal migration. Moreover, active Rac1 negatively regulates Rho or ROCK signaling and suppresses amoeboid movement. On the other hand, active Rho or ROCK supports bleb-based amoeboid migration23,45,49,51,52 and limits excessive Rac1-dependent adhesion via regulation of the Rac GAPs ARHGAP22 and filamin-A-associated Rho GTPase activation protein (FilGAP)23,53. Furthermore, cancer cells control amoeboid migration at the transcriptional level under circumstances in which matrix compliance allows sustained actomyosin contractility (Figure 1). Different chemical cues have been shown to control this process. For instance, amoeboid melanoma cells support contractility, establishing a positive feedback loop with the cytokines leukemia inhibitory factor (LIF)/IL6 and the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway to maintain Rho-ROCK activity49. As a result of high STAT3 activity, very contractile cells secrete different factors, including matrix metalloprotease 9 (MMP-9). MMP-9 promotes the generation of actomyosin contractile force and bleb-driven migration through a positive feedback loop via CD44 binding and increased MLC2 phosphorylation to sustain amoeboid invasion48. Moreover, amoeboid contractile cells secrete high levels of transforming growth factor beta (TGFβ), and downstream of it a Sma- and Mad-related protein 2 (SMAD2)-Cbp/P300-interacting transactivator with Glu/Asp-rich carboxy-terminal domain (CITED1) transcriptional network sustains actomyosin contractility54. In addition, the physical properties of the matrix play an important role in establishing a balance between actomyosin levels and adhesion to regulate optimal migration efficiency34,39,47,55,56. Increased ECM density results in increased matrix stiffness, in which cells sense and respond by increasing Rho-mediated actomyosin contractility57. Furthermore, slow mesenchymal cells can switch to fast amoeboid migrating modes under conditions of low adhesiveness and high physical confinement47,56.

ROCK-driven actomyosin contractility is stimulated by extracellular signals such as leukemia inhibitory factor (LIF) and transforming growth factor beta (TGFβ) to promote rounded amoeboid cancer cell motility. Rounded amoeboid cells display blebbing as well as high levels of actomyosin contractility and a rounded morphology. They interact with the extracellular matrix (ECM) by physically deforming it and by secreting metalloproteases (MMPs). In the stroma, ROCK-driven actomyosin contractility promotes the transformation of fibroblasts into cancer-associated fibroblasts (CAFs), driven by Yes-associated protein (YAP) as well as by extracellular factors. Blue indicates positive regulators of contractility, purple indicates negative regulators of contractility and orange lines indicate actomyosin contractility. Abbreviations: CAF, carcinoma-associated fibroblasts; CITED1, Cbp/P300-interacting transactivator with Glu/Asp-rich carboxy-terminal domain, 1; FilGAP, filamin-A-associated Rho GTPase activation protein; JAK, Janus kinase; RhoA, Ras homolog gene family member A; ROCK, Rho-associated protein kinase; SMAD2, Sma- and Mad-related protein 2; STAT3, signal transducer and activator of transcription 3.
The ability to switch between different modes of migration is an important factor for metastatic dissemination, as cancer cells have to migrate through a range of ECMs to escape the primary tumor and spread to distant organs. Therefore, anti-metastatic treatments should target the ability of tumor cells to cope with such variability. Recently, it has been described that potent ROCK inhibitors are able to strongly inhibit actomyosin contractility and collapse the actomyosin cytoskeleton, blocking both mesenchymal and amoeboid modes of movement32.
Intra-vital imaging studies have shown that bleb-driven highly contractile amoeboid migration is favored in the invasive fronts of melanomas and breast cancers23,29,45,48,49,58. Furthermore, in these studies, it has been shown that treatment with ROCK inhibitors or actomyosin perturbations (or both) is able to decrease tumor cell motility in vivo23,29,32,45,49,58. Hence, ROCK inhibition could effectively impair local invasion and dissemination of cancer cells (Figure 1).
Within the tumor, a variety of non-cancer stromal cells interact with the cancer cells promoting tumorigenesis7. Actomyosin contractility not only is fundamental for cancer cell migration and invasion but also is crucial for maintenance of the carcinoma-associated fibroblasts (CAFs) phenotype, an important stromal component in the tumor microenvironment7. Actomyosin contractility activated by ROCK signaling and the LIF/JAK/STAT pathway is crucial for CAF-dependent pro-invasive physical remodeling of the ECM favoring tumor aggressiveness and dissemination42,49,59,60. Additionally, actomyosin contractility, Src function, and matrix stiffening induced by TGFβ, are required for Yes-associated protein (YAP) activation in CAFs to promote ECM remodeling and cancer cell invasion, and to generate a positive feedback loop that helps to maintain the CAF phenotype61 (Figure 1). Moreover, contractility in CAFs has been shown to modulate EMT and metastasis-initiating cell properties in breast cancer models62.
Therefore, some degree of actomyosin contractility is essential for both cancer cells and stroma for efficient cell movement in the initial steps of the metastatic cascade34,41,49,59,61, and some factors such as TGFβ and LIF can stimulate contractility both in cancer cells and in fibroblasts.
After local invasion within the primary tumor microenvironment, cancer cells need to spread throughout the body and colonize new organs to form metastases. They do so by exploiting the vascular and lymphatic systems. The process through which cancer cells enter and exit vessels crossing the endothelial layer is known as transendothelial migration, which is extremely complex and involves the interaction with several different cell types, such as platelets, immune cells and endothelial cells, and the activation of a variety of signaling pathways63. These events are in some cases similar to those occurring during inflammation or infection, when immune cells need to enter and exit vessels. In fact, parallels between cancer cell and immune cell migration allow for interesting speculation in areas of cancer cell dissemination that are still not fully understood.
Intravasation. The first step in this metastatic cascade is intravasation, the entry of tumor cells into blood vessels. Intravasation depends on the weakening of cell-cell junctions between endothelial cells, which allows cancer cells to squeeze in between adjacent endothelial cells and enter the vessel lumen63. From a molecular perspective, not as much is known about intravasation compared with other steps in the metastatic cascade as this is an experimentally challenging step to study64,65. In fact, intravasation is dependent on the ability of cancer cells to invade towards blood vessels, so it is difficult to distinguish between genes involved in invasion and intravasation63. RhoA signaling has been linked to the process of intravasation66 (Figure 2). Specifically, RhoA activity in cancer cells is thought to be stimulated by macrophage contact and leads to the formation of invadopodia. Invadopodia are instrumental in the degradation and eventual breakdown of the matrix barrier, which allows for tumor cell intravasation. Furthermore, highly contractile, rounded amoeboid melanoma cells have been shown to intravasate more efficiently than low-contractility elongated cells in vivo67,68. Once in the bloodstream, cells are transported throughout the body by the blood flow (Figure 2).
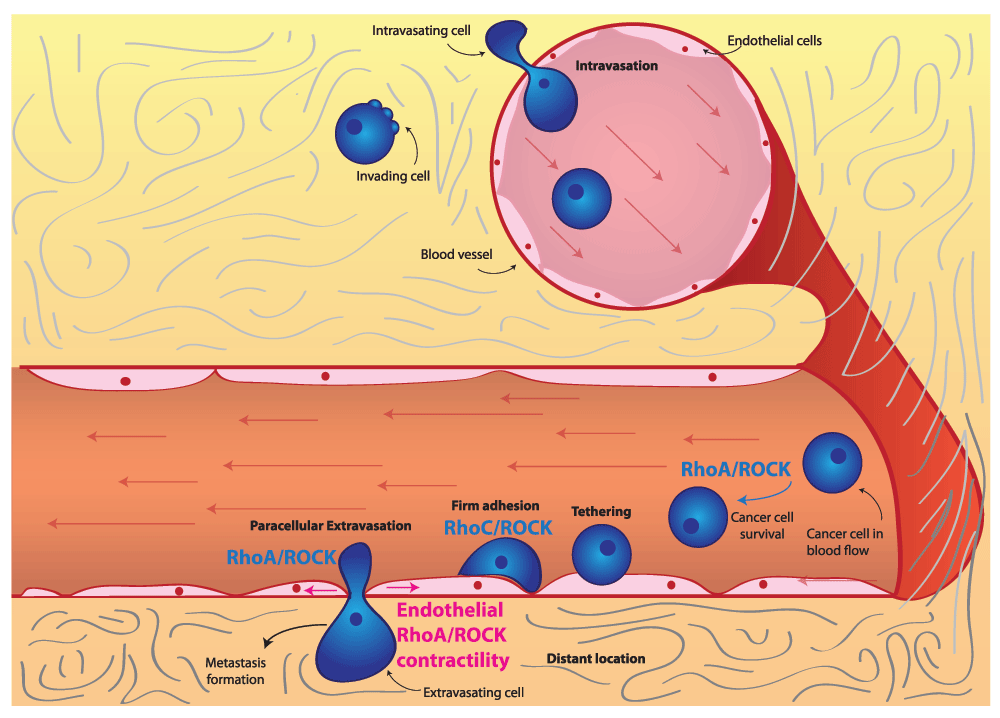
RhoC/ROCK signaling promotes survival of cancer cells in the blood flow as well as adhesion to the endothelium and extravasation. ROCK-driven actomyosin contractility within endothelial cells can be stimulated by secreted factors and is essential for cancer cell extravasation. Abbreviations: RhoC, Ras homolog gene family member C; ROCK, Rho-associated protein kinase.
Extravasation. Eventually, cancer cells flowing through the bloodstream need to exit blood vessels to form secondary tumors. This process is known as extravasation and entails several sequential steps. First of all, cancer cells form loose adhesions to the vascular endothelium, which is known as tethering. These loose adhesions then are tightened to form firm adhesions: firmly adhering cells then can cross the endothelial barrier and extravasate63.
The best-studied mechanism for extravasation is known as paracellular extravasation, during which cancer cells exit the vessel by squeezing in between endothelial cells. An alternative mechanism for cancer cell extravasation is transcellular extravasation, where tumor cells exit the vessel by going through endothelial cells63,69. Transcellular extravasation has been observed in immune cells70 and has also been identified in cancer cells, where it probably plays a role in some cases71.
Rho or ROCK-driven actomyosin contractility within extravasating cells has been shown to play an important role. For instance, in monocytes, RhoA activity has been shown to be necessary for tail retraction during extravasation72. In the context of transcellular extravasation, monocytes can rely on RhoA and ROCK signaling73,74.
On the other hand, in prostate cancer cells, it is RhoC and ROCK signaling that is essential for interaction with endothelial cells, promoting adhesion and paracellular extravasation75. As a result of its role in promoting extravasation, RhoC signaling is a key driver of tumor dissemination and metastasis75, in part explaining how RhoC was one of the first genes identified as a metastasis driver76. Furthermore, RhoA and RhoC have been shown to drive adhesion to the endothelium and transendothelial migration in breast and prostate cancer cells77,78. Consequently, rounded-amoeboid cancer cells with high levels of RhoA or ROCK-driven actomyosin contractility are more efficient during transendothelial migration than elongated cells both in vitro and in vivo67,68,79. Additional evidence supporting the importance of RhoA-driven contractility in transendothelial migration comes from studies examining the role of RhoA regulators. For instance, FilGAP, a Rac GAP, promotes RhoA signaling and rounded-amoeboid motility by suppressing Rac, and as a consequence it enhances in vivo extravasation of breast cancer cells53. Conversely, the RhoA GAP ARHGAP7 has been shown to be a negative regulator of transendothelial migration in thymic lymphoma80.
Cancer cells that successfully extravasate need to cross the vascular basement membrane that surrounds the vessel63. Since actomyosin contractility has been shown to promote the secretion of proteases in rounded amoeboid cells48, it is tempting to speculate that highly contractile extravasating cells could have an advantage when crossing the vascular basement membrane.
In order for paracellular extravasation to occur, cancer cells need to weaken cell-cell junctions within the endothelium. This can be mediated by regulating Rho or ROCK signaling and actomyosin contractility within the endothelial cells themselves (Figure 2). Lung cancer cells have been shown to induce adherens junction disassembly by stimulating actomyosin contractility through Rho/ROCK in endothelial cells81. Furthermore, thrombin stimulation of endothelial cells has been shown to induce ROCK activity and subsequently lead to cytoskeletal remodeling, junction disruption, and endothelial permeability82,83. Tumor-derived thrombin induces endothelial gap formation and transendothelial migration84. Furthermore, cancer cells have been shown to use thrombin within blood vessels in order to promote metastasis85. This prompts the speculation that actomyosin contraction in endothelial cells could be controlled by thrombin produced by cancer cells.
As well as leading to junction disassembly, actomyosin contractility in endothelial cells allows for endothelial cell retraction86,87, which increases endothelial permeability. Moreover, ROCK-driven actomyosin contractility in endothelial cells has been shown to prevent endothelial cell re-spreading downstream of ephrin-B signaling, which maintains increased endothelial permeability88. Conversely, ROCK inhibition has been shown to decrease endothelial permeability after hemorrhage89,90. Although these studies have not been conducted in cancer models, ROCK activity in endothelial cells could be similarly regulated while in contact with disseminating cancer cells.
In brief, we speculate that the ability of cancer cells to form secondary tumors is to a certain extent dependent on their ability to manipulate the cytoskeleton of endothelial cells; thus, increasing endothelial permeability could be a crucial step to promote extravasation. More work is needed to validate the roles of Rho/ROCK or actomyosin contractility (or both) in tumor cells during both cancer intravasation and extravasation.
Following extravasation at secondary sites, cancer cells that survive can form micro-metastasis and colonize new sites. In order for this colonization to take place, cancer cells must be able to adhere to endothelial cells, extravasate, survive and proliferate at the secondary site. The first few hours of colonization are crucial in determining the success of this process, as cells will undergo apoptosis if they do not adhere to their new niche. Furthermore, once established, cells must be able to evade the immune response in order to survive91. Although we have discussed that Rho/ROCK signaling is important for early dissemination, there is also evidence to suggest that Rho/ROCK signaling, actomyosin contractility or its regulators, or a combination of these are important for efficient colonization at secondary sites.
In vivo studies where cancer cells are injected intravenously (i.e., experimental metastasis assays) show that high levels of actomyosin contractility play a role in seeding of and colonizing the lung. For instance, cells selected for efficient colonization in the lung such as the highly metastatic A375M2 melanoma cell line have higher levels of RhoC76, RhoA23 and phosphorylated MLC248 when compared with low metastatic A375P melanoma cells.
Several studies have confirmed the importance of the initial hours in seeding during colonization. For example, serum response factor (SRF) co-activators myocardin-related transcription factors (MRTFs) are able to control the expression of MLC292 (Figure 3). MRTF and SRF are both important for early stages of lung colonization in breast cancer and melanoma92. Furthermore, depletion of MLC2 itself has also been shown to reduce lung colonization92. Conversely, enhanced actomyosin contractility favors colonization: for example, depletion of the actomyosin contractility suppressors Rac1 and its GEF dedicator of cytokinesis 3 (DOCK3) favors early lung colonization23. In melanoma, pigment epithelium-derived factor (PEDF) reduces lung colonization and suppresses lung tumor outgrowth93,94. PEDF is a negative regulator of Rho-ROCK signaling through supporting DOCK3-Rac1 activity95 (Figure 3). Furthermore, oncogenic BRAF suppresses phosphodiesterase 5A (PDE5A), which in turn inhibits actomyosin contractility96 (Figure 3). Therefore, re-expression of PDE5A reduces the ability of melanoma cells to colonize the lung and prevents short-term survival and long-term cancer growth in the lung96.
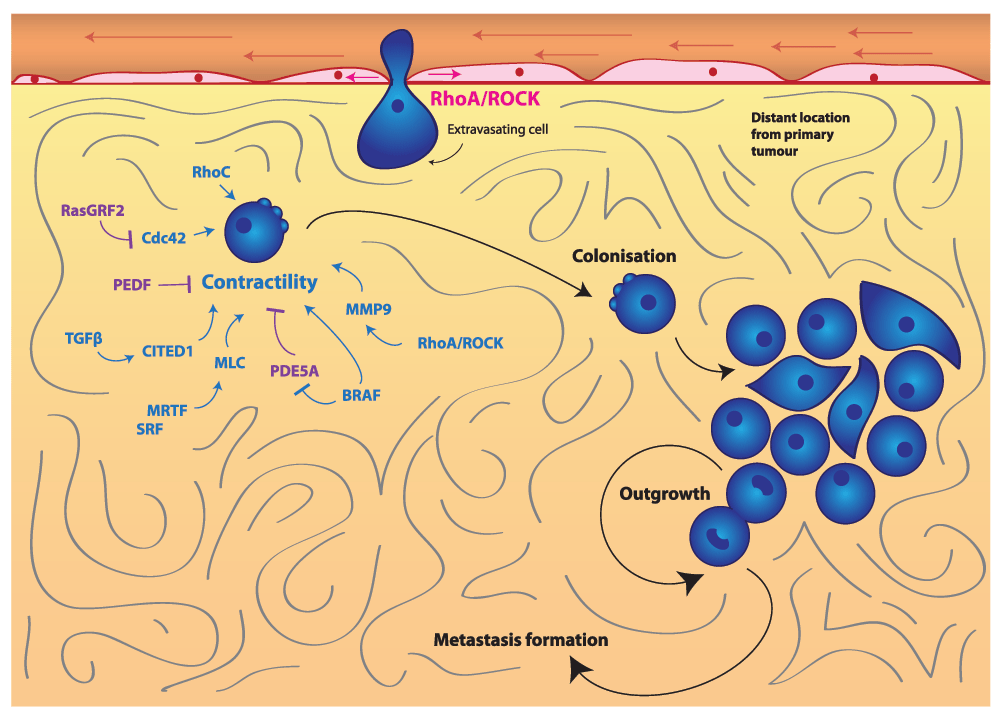
Actomyosin contractility promotes cancer cell colonization and outgrowth at a secondary site to form metastases. Contractility is under the control of a wide variety of pathways, including SRF/MRTF, TGFβ-SMAD-CITED1, MMP-9, BRAF-V600E and Cdc42 signaling. Blue indicates positive regulators of contractility, and purple indicates negative regulators of contractility. Abbreviations: CITED1, Cbp/P300-interacting transactivator with Glu/Asp-rich carboxy-terminal domain, 1; Cdc42, cell division control protein 42 homolog; MLC2, myosin light chain 2; MMP, matrix metallopeptidase; MRTF, myocardin-related transcription factors; PDE5A, phosphodiesterase 5A; PEDF, pigment epithelium-derived factor; RasGRF2, Ras protein-specific guanine nucleotide-releasing factor 2; ROCK, Rho-associated protein kinase; SMAD, Sma- and Mad-related protein; SRF, serum response factor; TGFβ, transforming growth factor beta.
As mentioned earlier, Cdc42 can also promote actomyosin contractility in cancer cells26. Further evidence of the importance of actomyosin contractility in early colonization has been shown by experiments in which loss of Ras protein-specific guanine nucleotide-releasing factor 2 (RasGRF2), an inhibitor of Cdc4297, enhanced colonization of the lungs in a Rac-independent manner. This was associated with higher actomyosin contractility levels97 (Figure 3).
TGFβ signaling plays an important role in promoting cancer cell colonization40,54,98 (Figure 3). We recently found that TGFβ increases actomyosin contractility in melanoma cells54. While TGFβ is known to promote EMT99 in epithelial cancers, in melanoma TGFβ signals through SMAD2 and the adaptor CITED1 to support contractile amoeboid migration54. TGFβ no longer sustains lung colonization in melanoma cells if the SMAD2-CITED1 axis is not functional54, which serves to highlight the multiple levels in which actomyosin contractility promotes colonization.
Furthermore, ROCK regulates expression of several MMPs, including MMP-9, which promote early stages of lung colonization48 (Figure 3). While MMPs exert their catalytic function in degradation of the ECM during local invasion, the non-catalytic roles of MMP-9 could promote the survival of cancer cells at the metastatic secondary sites. For example, it has been shown that non-catalytic functions of MMP-9 regulate STAT3 functions to drive survival in B-cell chronic lymphocytic leukemia (B-CLL) cells100.
From these results, it is clear that positive and negative regulators of Rho/ROCK signaling or actomyosin contractility (or both) are critical for cancer cells to efficiently colonize the metastatic sites in experimental metastasis models.
We have highlighted the crucial role that Rho/ROCK signaling or actomyosin contractility play in dissemination and metastatic colonization using a range of experimental cancer models. A highly contractile phenotype is clearly critical for effective cancer colonization, ultimately supporting the idea of developing drugs to inhibit actomyosin contractility. In vivo validation of the role of Rho/ROCK signaling or actomyosin contractility (or both) in metastasis is important to qualify these signaling modules as potential drug targets. Experimental metastasis models are insightful for understanding the processes of extravasation and colonization to the lungs, but recapitulation of the entire metastatic cascade, including local invasion, dissemination and intravasation, requires the use of spontaneous metastasis models101. Indeed, it has recently been shown that a new class of ROCK inhibitors has the ability to prevent both experimental and spontaneous metastases formation32. It will be of great importance to combine these mouse models with non-invasive cell-tracking techniques102,103 to understand the entire process and how early Rho/ROCK signaling should be targeted in order to effectively block the metastatic cascade.
CAF, carcinoma-associated fibroblasts; CITED1, Cbp/P300-interacting transactivator with Glu/Asp-rich carboxy-terminal domain, 1; Cdc42, cell division control protein 42 homolog; DOCK3, dedicator of cytokinesis 3; ECM, extracellular matrix; EMT, epithelial-mesenchymal transition; FilGAP, filamin-A-associated Rho GTPase activation protein; GEF, guanine nucleotide exchange factor; GAP, GTPase activation protein; GTP, guanosine triphosphate; JAK, Janus kinase; LIF, leukemia inhibitory factor; MLC2, myosin light chain 2; MMP, matrix metallopeptidase; MRTF, myocardin-related transcription factors; PDE5A, phosphodiesterase 5A; PEDF, pigment epithelium-derived factor; Rac1, Ras-related C3 botulinum toxin substrate 1; RasGRF2, Ras protein-specific guanine nucleotide-releasing factor 2; RhoA, Ras homolog gene family member A; RhoC, Ras homolog gene family member C; ROCK, Rho-associated protein kinase; SMAD2, Sma- and Mad-related protein 2; SRF, serum response factor; STAT, signal transducer and activator of transcription; TGFβ, transforming growth factor beta.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)