Keywords
RNA-seq, SAM/BAM alignments, education, cloud, hackathon, pipeline, workflow, alignment
This article is included in the Hackathons collection.
RNA-seq, SAM/BAM alignments, education, cloud, hackathon, pipeline, workflow, alignment
With the rise of RNA-seq for exploring biological hypotheses has come an increase in the number of algorithms for aligning RNA-sequences to the genome. The burden of selecting and properly using these algorithms often falls on biologists. However, many biologists do not have training or experience in the skills needed to select the proper tools. To provide an introduction for this audience, the Educational Environment Team sought to develop an interactive learning environment where students with a novice’s background in Unix could follow a series of alignment pipelines step-by-step.
The team identified two major goals that could be undertaken within the scope of the hackathon. The first was to produce a user-friendly tutorial that would walk novice computational biologists through the process of aligning RNA-seq data to the genome. The second was to supplement the tutorial with novel methods to compare the results of different RNA-alignment mappers.
Implementing this tutorial on an Amazon Machine Image (AMI) -- a virtual server in the cloud, pre-programmed with all the necessary packages -- allows students to initially bypass the intimidating task of installing software and dependencies, and immediately start performing alignments using a panel of different algorithms. All tasks in the tutorial and the AMI are designed to fall within the $35 per student provided for free through the Amazon Educate Program (http://aws.amazon.com/education/awseducate). By allowing students to run these alignments in succession, we hope to naturally showcase how these aligners vary in their outputs. Here we introduce both a read-based and position-based approach for identifying and evaluating regions of differential performance across the genome.
The team was composed of five members ranging in experience from graduate student to research faculty member. Each member of the team shared the experience of interfacing with non-computational biologists during their daily work and sought to provide a welcoming introduction to mapping tools. All members contributed to the goals of the team, with each selecting tasks to tackle according to their strengths.
As part of the tutorial, two team members dedicated their time to developing novel methods for comparing the results of RNA-alignment. After each of the town-hall style meetings conducted with the larger hackathon group, we received recommendations from members of other teams for software packages to help streamline the proposed analysis.
The tutorial was constructed as a GitHub Pages wiki -- each new wiki page represents a new step in the workflow1. We guide students through registration with the Amazon Educate Program, obtainment of data, alignment, and comparison of the performance of four different aligners. The comparison of aligners is achieved with our own custom written program, bamDiff, in concert with the R (v3.2.1) packages edgeR (v3.10) and csaw (v1.2.1).
The example dataset is sample NA12878 from the Genome in a Bottle Consortium, an extensively curated human-standard2. To give a flavor for the alignment workflow without burdening the learner with expensive computational requirements, we limited the genome for alignment to chromosome 20 of the latest human genome reference from RefSeq, GRCh38 (GCF_000001405.30). For the tutorial, we describe the use of four popular aligners: BWA v0.7.12-r10393, HISAT v0.1.6-beta4, STAR v2.4.0j5, and BLASTmapper (in preparation). For each, we provide the commands needed to construct an index and align the given data, outlining the expected screen output, files to be created, and time required for each task.
The R package csaw provides an elegant framework for identifying regions of differential expression between RNA-seq experiments, and in this case was extended to identifying regions of differential alignment between mappers6. To implement read counting, we used csaw to bin reads into 1KB windows, then filtered windows by count size7. Only bins with greater than 10 reads were kept. Windows were then filtered by exons, and only windows/bins containing exons were kept for further analysis (see Figure 1c). edgeR was used to identify windows/regions/bins with significant differences between the four mappers (see Figure 1d)8.
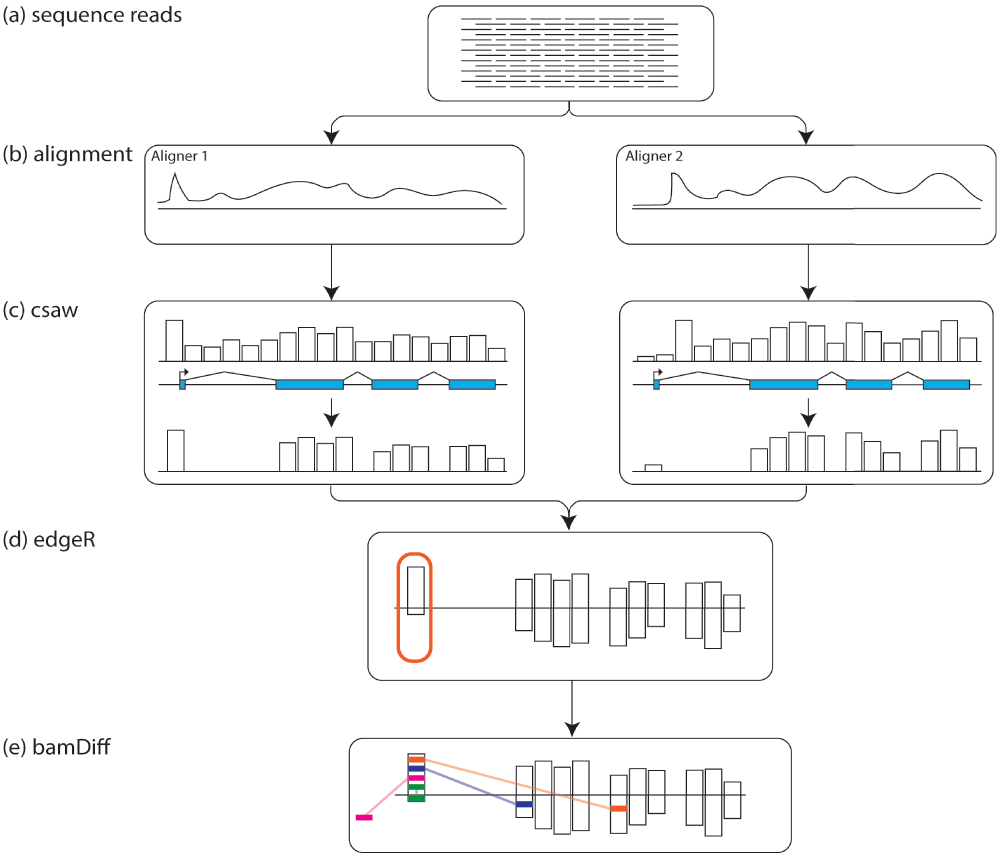
(a) Raw RNA sequencing reads undergo quality control checks and filtering before (b) being aligned using several different aligners. (c) csaw bins the reads and filters for bins based on read counts and overlap with exons. (d) edgeR identifies regions of where the aligners show differential mapping in the csaw filtered regions. (e) from the edgeR identified regions, bamDiff checks where those reads are being mapped in the other BAM files.
In order to systematically compare the performance of the various aligners, we wrote a new program called bamDiff. This Python script takes the output from csaw and edgeR as a CSV file as well as the outputs from each aligner in BAM format. bamDiff will report simple summaries for each BAM file, such as the total number of reads, overall number of alignments, proportion of reported reads that were unmapped by the aligner, as well as proportion of aligned reads that were mapped only once (uniquely).
The real strength of bamDiff comes in its ability to go beyond simple summarization to direct comparisons between BAM files. Internally bamDiff uses SAMtools9 to rapidly extract only the reads mapping to a region of interest identified by csaw from one BAM file. These reads are then checked against the other BAM files to see whether they are mapped at all, and if so, whether they are mapped to the same region in the genome as in the first BAM file (see Figure 1e). If they map to a conflicting region outside the region of interest, bamDiff will report the top ten regions reads are mapping to, by agglomerating reads mapping within 1kb of each other. These results are reported as text tables. Example usage and output can be viewed at the associated page in our tutorial: https://github.com/NCBI-Hackathons/RNA_mapping/wiki/7.-Compare-alignments-with-bamDiff.
Following the hackathon, our tutorial was integrated into NCBI NOW (National Center for Biotechnology Information Next Generation Sequencing (NGS) Online Workshop) a free online experience targeting individuals new to NGS analysis. The first offering of the workshop occurred October 13–23, 2015. Our tutorial was offered as a “take-home” exercise following the sixth lecture, which focused on the analysis of RNA-seq data.
We achieved two distinct goals during the hackathon: first, the development of a novel method for comparing the results of RNA-alignment and second, the creation of a tutorial to guide users through not only undertaking, but also understanding, RNA-seq alignment. Notably, we do not over-simplify the work of data analysis by simply selecting a single aligner for use in our tutorial. Instead, by encouraging the comparison of many different aligners we draw attention to the decision-making process intrinsic to any bioinformatic work. For our user, this is the selection of an RNA-seq aligner from many frustratingly similar options.
However, both the novel software bamDiff and the tutorial can be further improved.
Currently, bamDiff is best suited to analyzing a small region of the genome/BAM file, rather than undertaking the entire genome/BAM file. Additionally, a graphical summary would make outputs easier to understand.
In the tutorial we assume prior knowledge of the contents, purpose, and format of FASTQ, SAM, and BAM files as we assumed that our tutorial would be used in conjunction with a class or workshop that covers these introductory topics, such as NCBI NOW. The tutorial could be expanded in a number of ways, such as generalizing to pair-end reads or alignments guided by an annotation. Finally, there may be room for improvements in usability, such as streamlining use on platforms such as Google Cloud, Microsoft Azure, or iPlant/CyVerse.
Latest source code: https://github.com/NCBI-Hackathons/RNA_mapping.
Source code as at the time of publication: http://dx.doi.org/10.5281/zenodo.5087110
License: CC0 1.0 Universal
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)