Introduction
Visualizing proteins indirectly in cells and tissues at the light microscopic level using antibodies conjugated to fluorochromes revolutionized the field of cell biology 40 years ago. In the late 1980s and early 1990s, the use of commercial, user-friendly, confocal microscopes in combination with digital image acquisition systems dramatically improved the image quality of fluorescently labeled specimens and made image capture easier by avoiding the vagaries of the dark room. The technical benefits of confocal over “conventional” microscopy include the removal of the out-of-focus glare that interferes with what the imager really wishes to observe and an increase in specimen contrast. More recently, the ability to follow tagged molecules in live cells using both conventional and confocal fluorescence microscopy and the development of molecular biosensors has allowed cell biologists to study the dynamics of individual proteins and protein complexes with high precision. Conventional techniques used for light microscopy can achieve resolutions of up to ~100 nm in the image plane, as first detailed by Abbe1, and about twice this value along the focal axis2. In practice, most authors consider the resolution of commercially available microscopes to be ~200 nm in the image plane and ~500 nm in the axial dimension. So called “super resolution” is achieved with techniques that allow resolution beyond the diffraction limit of conventional optics3. The term super resolution as applied to microscopy has been in use since at least as early as the 1960s4. Resonance energy transfer5, near field scanning optical microscopy6, dual objective (4Pi) microscopy7, total internal reflection microscopy8, single molecule fluorescence localization9, expansion microscopy10, and several other strategies represent successful efforts to obtain structural or positional information from biological specimens at resolutions better than those afforded by conventional microscopy. However, many of these approaches are technically demanding or present significant limits to the type of specimens that can be examined. The last decade has seen the development of methods that closely resemble more familiar far-field and laser scanning confocal microscopy but allow direct visualization of subdiffraction size structures in fixed and living specimens. Current super resolution techniques provide resolutions of less than 10 nm in the image plane11 and ~20 nm in the z-axis. The improved resolution offered by these methods has produced breathtaking images of the nuclear pore complex12,13, the tubular walls of microtubules14, and many other structures.
Although the potential and allure of super resolution methods are indisputable, they also present new challenges to image acquisition, storage, and interpretation. For example, changing the resolution of an image from 200 nanometers to 20 nanometers in the image (xy) plane, while maintaining a fixed field of view, results in a 100-fold increase in image size. Extending these calculations to the third dimension, multispectral imaging and time (for live cells) reveals the potential extent of demands that super resolution microscopy can place on specimens and fluorophores as well as imaging and data processing hardware. Moreover, although computer and imaging technologies have advanced to the point where accumulating these data is feasible, human involvement is currently still needed to identify regions of interest for examination and analysis. Each of the methods used to achieve super resolution imaging also offer unique technical strengths and weaknesses to cell biologists. We direct the reader to several excellent recent reviews that provide an overview of the capabilities, advantages, and disadvantages of a variety of super resolution procedures in tabular form15–18.
Despite the inherent challenges, the use of super resolution microscopy is beginning to make an impact on a wide variety of biological topics. Among the subjects most likely to benefit from the application of super resolution imaging is the study of the cytoskeleton and its associated structures. Unlike many other cellular components, cytoskeletal filaments form anastomosing networks of fibers smaller than the resolution of conventional imaging methods. Techniques allowing enhanced resolution imaging of cytoskeletal structures, especially in live cells, are already advancing our knowledge of their formation and function. For example, super resolution has been a boon to investigators studying cytoskeletal rearrangements in bacteria19,20 and yeast cells21, which have generally been too small to approach with conventional diffraction-limited imaging methods. Below, we briefly discuss the major methods for achieving super resolution as well as their strengths and weaknesses with emphasis on recent work in which these methods have been applied to address biological questions involving the cytoskeleton.
Localization microscopy
Stochastic optical reconstruction microscopy (STORM22), photoactivated localization microscopy (PALM23), fluorescence photoactivation localization microscopy (FPALM24), and a growing number of related methods are techniques where fluorescent specimens are examined by activating a limited set of fluorophores at a time which must be separated by distances greater than the resolution limit of the microscope. A diffraction-limited image of the fluorophores is captured and the position and intensity of each fluorophore calculated at subdiffraction precision. Activated fluorophores are deactivated, and a new set of fluorophores is activated and imaged. After many images are collected, a completed super resolution image is calculated. The techniques differ in the type of fluorophore used. For example, PALM and FPALM are used to image expressed photoactivatable fluorescent proteins and fusion proteins23,24, while STORM is used to create images of fluorescent dyes and tags that can be switched between fluorescing and non-fluorescing states22. In practice, these methods can produce the highest resolution of the available diffraction-unlimited techniques when applied to biological specimens, with two-dimensional resolutions of less than 20 nm frequently reported12,25–27. However, overall image resolution and quality increase with the number of photons captured and fluorescent molecules examined. Therefore, these methods are limited by the time required to obtain a sufficient number of images – often tens of thousands – needed to create a final super resolution image. In addition, many conventional fluorophores and fluorescent proteins are not suitable for localization microscopy14,28, making some techniques such as multicolor staining methods challenging. Localization methods also require that fluorescent molecules or proteins within the specimen be detected individually, complicating the application of these techniques to densely labeled three-dimensional cytoskeletal arrays or arrays contained within thick samples exhibiting autofluorescence. To address this, some investigators have incorporated total internal reflection fluorescence illumination (TIRF), two photon illumination, or light sheet illumination to limit the volume of a specimen under inspection23,29–31. However, these implementations increase the complexity of the instrumentation required. Despite these limitations, a large number of studies encountered in our review of current literature employ localization techniques. This may be because of both the superior resolution of the methods and the relatively simple hardware requirements which have allowed many investigators to build their own STORM or PALM/FPALM imaging systems. In addition, fluorophores, fluorescent proteins, buffering agents, and illumination strategies are being actively developed to extend localization to both multicolor labeling and other fluorescent imaging tasks (see 28,32–35 for representative reviews).
Among the most novel and dramatic discoveries made using localization methods is the periodic distribution of actin filaments and associated cytoskeletal proteins in axons first visualized using STORM imaging36. The periodicity of this structural feature of neurons is below the resolution limit of conventional microscopy and was overlooked in thin section electron micrographs37,38. Other studies have further exploited single molecule/protein imaging to analyze the development and regulation of these arrays39,40. STORM and PALM methods are also being applied to the study of the structure of adhesion complexes such as hemidesmosomes41, intercellular adherens junctions42,43, and the less well-defined adhesions formed by leucocytes44, as well as actin cytoskeletal rearrangements that occur during endocytosis45 and bacterial host cell invasion46. In the case of adherens junctions, STORM analyses indicate that E-cadherin exists in clusters at sites of cell-cell interaction rather than as the “solid” belt typically observed by non-super resolution methods42,43. Similarly, dual-color PALM studies suggest that paxillin and vinculin form functionally distinct non-overlapping nanoaggregates in focal adhesions that are not detectable using conventional imaging methods47.
Microtubules, 24 nm diameter tubes composed of protofilaments, are sparsely distributed at the edge of cultured cells and are often used as proof-of-concept targets by developers of super resolution imaging methods48–51. STORM and PALM imaging are beginning to provide new details regarding the function and organization of the microtubule cytoskeleton. For example, these techniques have been used to study the organization of centrosomal proteins in intact cells, the architecture of microtubules underlying the movement of organelles within living cells52, and the interaction of kinesin motor proteins with microtubules in neuronal processes53. A variant of PALM imaging has also been used to study the structure of EB1 at the distal tip of growing microtubules54, and PALM has made possible the visualization of FtsZ, the bacterial homolog of eukaryotic tubulin, in distinct polymeric arrays in prokaryotes20,55.
Although relatively few studies have examined intermediate filament arrays using super resolution imaging, STORM has been used to investigate keratin, plectin, and integrins in hemidesmosomes formed by cultured keratinocytes41. Additionally, desmin, a cytoskeletal protein mutated in clinically important cardiomyopathies, has been visualized using dual color PALM microscopy in cultured cardiomyocytes56. In the latter study, the authors report a 10-fold increase in the resolution of desmin protein aggregates and filaments over non-super resolution light microscopic methods. More importantly, their super resolution images reveal that both mutant and wild-type desmin proteins are incorporated into the same filament, suggesting the possibility that changes in the mechanical properties of a “mixed” filament might be the cause of disease56.
Structured illumination microscopy
Structured illumination microscopy (SIM) improves the resolution of light microscopy by illuminating a specimen with a defined regular pattern of diffraction-limited light and dark bands which create Moiré patterns when combined with the structure of a specimen57. An image of the resulting interference pattern is created and recorded. The illuminating pattern is then rotated and further images captured. Finally, an image with improved resolution is calculated from the combined rotation series. Because the illuminating pattern is generated using wide-field optics, the technique exposes a specimen to illumination levels that are comparable to other wide-field microscopy methods (although multiple images must be captured for each view of the specimen). While many implementations of localization microscopy use total internal reflection illumination and are therefore limited to observation of structures within ~100 nm of an optical surface, SIM can resolve structures many microns deep within a specimen. SIM can also be used with any fluorescent probe and, unlike localization methods or laser scanning methods such as stimulated emission depletion microscopy (STED), complete images of a specimen are obtained at speeds determined by camera sensitivities. SIM is therefore one of the least phototoxic and most rapid methods for obtaining enhanced resolution images and has been used extensively in studies of living cells. A three-dimensional version of structured illumination allows for improved resolution in the z direction and has been used to visualize cytoskeletal structures in three dimensions. Both of these applications are discussed below.
The resolution achieved with SIM is generally only twofold greater than that offered by conventional microscopy57, although some implementations allow SIM to achieve lateral resolutions as high as 50 nm58,59. However, even a twofold increase in resolution, combined with the power of multispectral fluorescent labeling methods, has allowed investigators to observe previously unresolved features of a wide variety of cytoskeletal structures. For example, SIM has been used to characterize the distribution of microtubules and associated structures in a variety of specimens that conventional diffraction-limited imaging methods have been unable to resolve well, including the neuromuscular junction60, platelets61, centrosomes62,63, and protists64. SIM has also facilitated studies investigating the structure of striated muscle65,66 as well as actin and myosin filament organization in non-muscle cells67–70. Intermediate filament architecture and associated junctions have also been examined using this method71,72. Interestingly, although the localization-based imaging methods described above achieve higher resolution than SIM, the pointillized appearance of images produced by localization microscopy can obscure fine structural detail that may be visible using SIM. For example, SIM has revealed that focal adhesions comprise linear subarrays73, a feature not readily visible in images published by investigators using interferometric (i)PALM to analyze the axial distribution of focal adhesion components74–76.
Finally, because SIM can be applied to microscopy of any fluorescent probe, it can be readily used for multispectral studies using conventional fluorophores. SIM has been used to examine cytoskeletal structures in triple-labeling studies of adherens junctions77,78, neuronal spines79, centrioles80, kinetochores81, and endosomal vesicles82. For example, SIM analyses of VASP, zyxin, testin, and other proteins surprisingly indicate that these tension-regulating proteins are likely recruited to adhesion junctions independent of the core adhesion complex78.
Stimulated emission depletion microscopy
STED, reversible saturable optical fluorescence transitions microscopy (RESOLFT), and related techniques excite fluorophores in a diffraction-limited spot by a focused laser. Outlying fluorophores are converted to a non-fluorescent state by illumination with a second (depletion) laser in a manner that leaves a central, subdiffraction limited area of fluorophores unconverted83–85. The remaining still-fluorescent fluorophores are detected to create an image with resolutions far greater than those provided by conventional imaging methods. Some implementations of these techniques have yielded resolutions in biological specimens of <50 nm in the image plane and 150 nm in the axial dimension86. Common implementations of STED are technically demanding and the depletion light energies are substantially greater than the illumination intensities required by other super resolution imaging methods. However, STED and similar systems closely resemble laser scanning confocal microscopes already employed by many investigators, and these methods can be applied to most commonly available fluorophores. Unlike localization methods and SIM, STED does not require calculations to generate enhanced resolution images, and image resolution can be readily varied by changing the raster scanning patterns used to visualize a specimen. Scanning rates achieved by STED are suitable for imaging of live specimens87,88, but the required depletion energies currently make extended live cell imaging a challenge. However, super resolution imaging can also be interchanged with conventional confocal scanning approaches simply by turning the depletion laser on or off.
STED has been used to visualize the periodic ring structure of actin in neurons89 and actin dynamics in dendritic spines of neurons in living brain tissue90. Others have used STED to examine the actin-like MreB protein in bacteria91 and the reorganization of actin filament arrays during activation of natural killer cells and T cells92,93. It has allowed the visualization of myosin mini-filament formation in mammalian non-muscle cells94 and insight into the regulation of the actin cytoskeleton by intracellular signaling proteins95,96. Microtubules have been examined using STED in primary cilia97, and the interphase microtubule array has been examined in muscle and non-muscle cells98,99. Finally, several groups have utilized STED microscopy to visualize elements of the intermediate filament network, including vimentin100,101, keratin102, and nuclear lamins103. In our laboratory, we have used STED to assay the relative localization of vimentin and focal adhesions in cultured epithelial cells. Whether vimentin interacts with focal adhesions has been controversial for years104,105. However, STED not only provides images with more detail than can be obtained using conventional confocal microscopy but also reveals a distinct pattern of organization of paxillin within a focal adhesion (Figure 1). Moreover, in the STED image, vimentin filaments clearly wrap around each focal adhesion, an interaction that is not apparent in the confocal image.
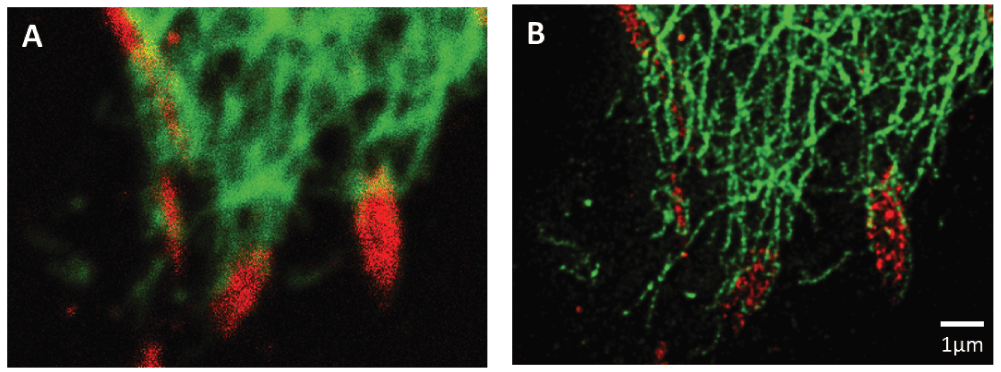
Figure 1. Association of paxillin (Alexafluor 555, red) and vimentin (Oregon green 488, green) at focal adhesions located at the leading edge of a migrating A549 lung cell.
Conventional confocal imaging is shown in A while STED imaging of the same area is shown in B.
Live cells
The ability to study proteins in cells in the living state is one of the most significant advantages of light microscopy over other methods of analysis. However, as has been noted by many others (see 106–108 for representative reviews), obtaining images of living cells that are doing something other than dying on the microscope is difficult and involves balancing the competing requirement of image quality and cell health. In addition, the study of cytoskeletal dynamics often requires rapid image acquisition, increasing the light exposure of live cells over time. Achieving resolutions greater than the diffraction limit of conventional microscopy ultimately requires capturing larger numbers of photons from smaller areas of live cells than is necessary for conventional resolution images, an effort that can clearly compromise the integrity of cells under examination108. Fluorescently labeled cytoskeletal filaments are also notoriously photolabile109. To date, many investigators of the cytoskeleton in live cells have combined super resolution microscopy of fixed cells with wide-field or confocal imaging of live cells39,44,45,52,53,64,68,69,77,79,98,110–113 or have obtained single images of live cells using super resolution methods56,89. Although the latter avoid potential artifacts induced by fixation and immunostaining, this approach does not address dynamic changes in cell architecture.
The majority of publications in our survey that examined dynamic changes in cytoskeletal architecture employed SIM. Observations of the cytoskeleton in live cells have been made over time scales ranging from seconds to tens of minutes using this approach. The twofold resolution enhancement offered by SIM has allowed investigators to examine in previously impossible detail the growth of microtubules in living plant cells114, actin retrograde flow in cultured insect cells115, and the reorganization of cytoskeletal arrays in dividing yeast and bacterial cells21,91. Two-color applications of SIM in living cells have been used to demonstrate heterotypic assembly of myosin II isoforms67, the spatial relationship between myosin IIA and alpha actinin116, the clustering of receptors in adherens junctions117, and that vimentin intermediate filaments move bi-directionally along microtubules118.
Although capturing clear images of closely apposed structures using localization methods requires accumulation and processing of thousands of images over periods of many seconds to minutes, localization methods have also been applied to the study of rapidly changing cytoskeletal structures in live cells by sacrificing some image clarity to improve temporal resolution. For example, PALM has been used to quantify the addition and loss of individual paxillin proteins at focal adhesion sites with a spatial resolution of 60 nm and temporal resolution of 25 seconds119. Proof-of-principle studies have also demonstrated that PALM can be used to visualize dynamic changes in fluorescent actin filaments over brief intervals120 and the movement of fluorescently labeled endosomes along microtubules in living axons121. In addition, PALM imaging platforms have been exploited by several groups that have followed changes in localization of single fluorescent cytoskeletal proteins over time, either alone or in combination with PALM, STORM, or STED techniques122–125. This appears to be a powerful, multimodal approach that can place the movement of individual proteins in the context of cytoskeletal architecture.
STED employs depletion lasers at energies that are largely incompatible with extended viewing of live cells, and, not surprisingly, we encountered few publications where this method has been used to image cytoskeletal arrays in vivo. Nonetheless, STED has great potential for imaging structures within complex tissues and has been used in a seminal study of actin dynamics in living neurons within brain tissue90. Single images of microtubules within living cells have also been obtained at 60 nm resolution using STED86. STED has also been applied to imaging using multiphoton excitation126–128 and total internal reflection microscopy129. These methods limit light exposure to a sample and offer further potential for the application of STED illumination to live cells. We anticipate that as these complex instruments become more widely available, we will see an increase in the number of investigators employing them for studies of living cells.
The third dimension
Many initial implementations of super resolution methods did not provide an increase in resolution in the third, or axial, dimension of the microscope (see 18 for review). However, recent modifications to these methods have achieved impressive resolution enhancements in the third dimension. Three-dimensional SIM doubles the resolving ability of the light microscope in all dimensions while retaining its ability to obtain images rapidly with low light exposures130. Although many studies have captured Z-stacks of images using super resolution methods and generated extended focus images from them, relatively few studies have analyzed the cytoskeleton in three dimensions using super resolution microscopy. However, three-dimensional SIM has been used to resolve actin filament arrays and microtubules in three dimensions using both fixed and live cultured cells113,116,130,131 as well as to study the structure of centrosomes63,80 and kinetochores81. Additionally, this method has also been used to visualize the three-dimensional organization of FtsZ in dividing bacteria132. Modification of STORM and PALM imaging platforms can achieve axial resolution of up to 20 nm by introducing axial astigmatism into the optical path133 and by generating interference patterns from images obtained with paired, opposed objective lenses134. Three-dimensional STORM and PALM have been used to study the movement of organelles along microtubules in live cells and the formation of FtsZ ring structures in live dividing bacteria20,52. A STORM imaging method employing axial astigmatism and dual objectives has been used to show that sheet-like cellular extensions in cultured cells contain two distinctly separate actin filament arrays each with unique patterns of actin filament organization134. The development and regulation of these previously undetected cytoskeletal arrays have been further analyzed using similar approaches in normal cell movement112,116. iPALM can resolve structures less than 20 nm in diameter in three dimensions27. The technique has recently been used to dissect the organization of cytoskeletal proteins associated with matrix adhesion devices termed focal adhesions at an unprecedented level of detail74–76. Impressively, the technique was able to resolve not only vertical stratification enriched for specific cytoskeletal components but also the polarized orientation of the N- and C-terminal ends of talin within focal adhesions of intact cells. Finally, STED microscopy has also been modified by the addition of dual depletion patterns, one oriented in the image plane and the other in the axial dimension, allowing for increases in resolution in the third dimension of up to 125 nm135. STED and RESOLFT have also been extended to the third dimension using a dual objective imaging strategy136.
Presently, most work using super resolution imaging has been conducted using single cells cultured on optical surfaces. However, previous work has shown that cell morphology and behavior can be dramatically altered in three-dimensional environments137,138. To date, the literature examining the cytoskeleton in cells in situ is largely limited to proof-of-principle studies. However, these efforts demonstrate the growing potential of super resolution methods. For example, STED has been used to view the dynamics of actin filament arrays within live neurons in 350 μM thick brain slices90, and three-dimensional SIM has been used to visualize actin arrays in developing Drosophila115. In other studies, labeled nuclear histones have been visualized in cells within 150 μM cell spheroids by combining single molecule localization methods with planar illumination139. Both planar and multi-photon illumination methods have been coupled with structured illumination to view green fluorescent protein (GFP) expressed in living nematodes140,141. These methods improve visualization by restricting effective illumination to a single plane of interest within thick specimens. While these latter studies examined structures other than the cytoskeleton, these methods show promise for the analysis of the cytoskeleton at subdiffraction resolutions in situ.
Summary
Many of us have been fortunate to work as cell biologists during two major revolutions in imaging technology: the development of fluorescent proteins as tools for biologists and the development of confocal microscopy, which extracts clear, in-focus images in which the contaminating blur of out-of-focus structures has been removed. The astonishment and wonder with which we now view images created by super resolution microscopy suggest that we are experiencing yet a third revolution in the technology available to cell biologists. It is likely that for dual and triple labeling studies of cultured cells in two and three dimensions, as well as studies of bright, relatively slow-moving structures in live cells, super resolution imaging will soon replace confocal microscopy as the state of the art in much the same way that confocal microscopy replaced conventional wide-field imaging in the 1980s and 1990s. However, each of the approaches used to achieve images at better than diffraction-limited resolution have strengths and weaknesses. Studies of rapid cytoskeletal dynamics in live cells and three-dimensional studies are likely to present technical and biological challenges to practitioners of super resolution microscopy for some time. In addition, while the best super resolution light microscopic methods achieve resolutions of <10 nm, this is still 50–100-fold greater than the resolution afforded by electron microscopy142,143. Conventional fluorescence microscopes can also take advantage of a myriad of probes for physiological conditions and molecular interactions that have yet to be adapted to super resolution imaging methods. For the time being, no method addresses all possible experimental needs, and investigators will likely have to address the limitations of their super resolution instruments with complementary approaches involving conventional methods. Moreover, the dream of seeing individual protein complexes and their partners functioning in live cells within a complex three-dimensional organism remains unrealized. Nonetheless, we are seeing the development of instrumentation, computational methods, fluorescent probes, and novel methods at an amazing pace. We can only imagine what the next advances will be.
Author contributions
Eric A. Shelden wrote the majority of the text with the assistance of Jonathan C.R. Jones. Jonathan C.R. Jones and Zachery T. Colburn edited the text. Zachery T. Colburn provided the images and processed cells for localization.
Competing interests
The authors declare that they have no competing interests.
Grant information
This work was supported by grants from the National Science Foundation (IOS 1457368) to EAS and National Institutes of Health (AR054184) to Jonathan C.R. Jones. Zachery T. Colburn was supported, in part, by a Poncin Scholarship.
Faculty Opinions recommendedReferences
- 1.
Abbe E:
Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung.
Archiv f mikrosk Anatomie.
1873; 9(1): 413–8. Publisher Full Text
- 2.
Francon M:
Phase-contrast microscopy. Progress in Microscopy. New York: Row, Peterson & Co, 1961.
- 3.
Kolobov MI:
Quantum Imaging. New York, NY: Springer New York; 2007. Publisher Full Text
- 4.
Lohmann AW, Paris DP:
Superresolution for Nonbirefringent Objects.
Appl Opt.
1964; 3(9): 1037. Publisher Full Text
- 5.
Stryer L:
Fluorescence energy transfer as a spectroscopic ruler.
Annu Rev Biochem.
1978; 47: 819–46. PubMed Abstract
| Publisher Full Text
- 6.
Pohl DW, Courjon D:
Near field optics. Springer Science & Business Media.
2012; 242.
- 7.
Hell S:
Double-scanning confocal microscope.
European patent.
1990; 491289.
- 8.
Axelrod D:
Cell-substrate contacts illuminated by total internal reflection fluorescence.
J Cell Biol.
1981; 89(1): 141–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 9.
Yildiz A, Forkey JN, McKinney SA, et al.:
Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization.
Science.
2003; 300(5628): 2061–5. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 10.
Chen F, Tillberg PW, Boyden ES:
Optical imaging. Expansion microscopy.
Science.
2015; 347(6221): 543–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 11.
Rittweger E, Han KY, Irvine SE, et al.:
STED microscopy reveals crystal colour centres with nanometric resolution.
Nature Photon.
2009; 3: 144–7. Publisher Full Text
- 12.
Löschberger A, van de Linde S, Dabauvalle MC, et al.:
Super-resolution imaging visualizes the eightfold symmetry of gp210 proteins around the nuclear pore complex and resolves the central channel with nanometer resolution.
J Cell Sci.
2012; 125(Pt 3): 570–5. PubMed Abstract
| Publisher Full Text
- 13.
Schermelleh L, Carlton PM, Haase S, et al.:
Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy.
Science.
2008; 320(5881): 1332–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 14.
Dempsey GT, Vaughan JC, Chen KH, et al.:
Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging.
Nat Methods.
2011; 8(12): 1027–36. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 15.
Fornasiero EF, Opazo F:
Super-resolution imaging for cell biologists: concepts, applications, current challenges and developments.
Bioessays.
2015; 37(4): 436–51. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 16.
Galbraith CG, Galbraith JA:
Super-resolution microscopy at a glance.
J Cell Sci.
2011; 124(Pt 10): 1607–11. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 17.
Godin AG, Lounis B, Cognet L:
Super-resolution microscopy approaches for live cell imaging.
Biophys J.
2014; 107(8): 1777–84. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 18.
Schermelleh L, Heintzmann R, Leonhardt H:
A guide to super-resolution fluorescence microscopy.
J Cell Biol.
2010; 190(2): 165–75. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 19.
Biteen JS, Goley ED, Shapiro L, et al.:
Three-dimensional super-resolution imaging of the midplane protein FtsZ in live Caulobacter crescentus cells using astigmatism.
Chemphyschem.
2012; 13(4): 1007–12. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 20.
Holden SJ, Pengo T, Meibom KL, et al.:
High throughput 3D super-resolution microscopy reveals Caulobacter crescentus in vivo Z-ring organization.
Proc Natl Acad Sci U S A.
2014; 111(12): 4566–71. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 21.
Dudin O, Bendezú FO, Groux R, et al.:
A formin-nucleated actin aster concentrates cell wall hydrolases for cell fusion in fission yeast.
J Cell Biol.
2015; 208(7): 897–911. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 22.
Rust MJ, Bates M, Zhuang X:
Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM).
Nat Methods.
2006; 3(10): 793–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 23.
Betzig E, Patterson GH, Sougrat R, et al.:
Imaging intracellular fluorescent proteins at nanometer resolution.
Science.
2006; 313(5793): 1642–5. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 24.
Hess ST, Girirajan TP, Mason MD:
Ultra-high resolution imaging by fluorescence photoactivation localization microscopy.
Biophys J.
2006; 91(11): 4258–72. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 25.
Greenfield D, McEvoy AL, Shroff H, et al.:
Self-organization of the Escherichia coli chemotaxis network imaged with super-resolution light microscopy.
PLoS Biol.
2009; 7(6): e1000137. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 26.
Jones SA, Shim SH, He J, et al.:
Fast, three-dimensional super-resolution imaging of live cells.
Nat Methods.
2011; 8(6): 499–508. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 27.
Shtengel G, Galbraith JA, Galbraith CG, et al.:
Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure.
Proc Natl Acad Sci U S A.
2009; 106(9): 3125–30. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 28.
Chozinski TJ, Gagnon LA, Vaughan JC:
Twinkle, twinkle little star: photoswitchable fluorophores for super-resolution imaging.
FEBS Lett.
2014; 588(19): 3603–12. PubMed Abstract
| Publisher Full Text
- 29.
Baddeley D, Crossman D, Rossberger S, et al.:
4D super-resolution microscopy with conventional fluorophores and single wavelength excitation in optically thick cells and tissues.
PLoS One.
2011; 6(5): e20645. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 30.
Chen BC, Legant WR, Wang K, et al.:
Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution.
Science.
2014; 346(6208): 1257998. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 31.
Cella Zanacchi F, Lavagnino Z, Faretta M, et al.:
Light-sheet confined super-resolution using two-photon photoactivation.
PLoS One.
2013; 8(7): e67667. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 32.
Lippincott-Schwartz J, Patterson GH:
Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging.
Trends Cell Biol.
2009; 19(11): 555–65. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 33.
Shcherbakova DM, Sengupta P, Lippincott-Schwartz J, et al.:
Photocontrollable fluorescent proteins for superresolution imaging.
Annu Rev Biophys.
2014; 43: 303–29. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 34.
Wiedenmann J, Gayda S, Adam V, et al.:
From EosFP to mIrisFP: structure-based development of advanced photoactivatable marker proteins of the GFP-family.
J Biophotonics.
2011; 4(6): 377–90. PubMed Abstract
| Publisher Full Text
- 35.
Wu B, Piatkevich KD, Lionnet T, et al.:
Modern fluorescent proteins and imaging technologies to study gene expression, nuclear localization, and dynamics.
Curr Opin Cell Biol.
2011; 23(3): 310–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 36.
Xu K, Zhong G, Zhuang X:
Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons.
Science.
2013; 339(6118): 452–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 37.
Jones SL, Korobova F, Svitkina T:
Axon initial segment cytoskeleton comprises a multiprotein submembranous coat containing sparse actin filaments.
J Cell Biol.
2014; 205(1): 67–81. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 38.
Watanabe K, Al-Bassam S, Miyazaki Y, et al.:
Networks of polarized actin filaments in the axon initial segment provide a mechanism for sorting axonal and dendritic proteins.
Cell Rep.
2012; 2(6): 1546–53. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 39.
Ganguly A, Tang Y, Wang L, et al.:
A dynamic formin-dependent deep F-actin network in axons.
J Cell Biol.
2015; 210(3): 401–17. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 40.
Zhong G, He J, Zhou R, et al.:
Developmental mechanism of the periodic membrane skeleton in axons.
eLife.
2014; 3: e04581. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 41.
Nahidiazar L, Kreft M, van den Broek B, et al.:
The molecular architecture of hemidesmosomes, as revealed with super-resolution microscopy.
J Cell Sci.
2015; 128(20): 3714–9. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 42.
Erami Z, Timpson P, Yao W, et al.:
There are four dynamically and functionally distinct populations of E-cadherin in cell junctions.
Biol Open.
2015; 4(11): 1481–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 43.
Wu Y, Kanchanawong P, Zaidel-Bar R:
Actin-delimited adhesion-independent clustering of E-cadherin forms the nanoscale building blocks of adherens junctions.
Dev Cell.
2015; 32(2): 139–54. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 44.
Wee JL, Schulze KE, Jones EL, et al.:
Tetraspanin CD37 Regulates β2 Integrin-Mediated Adhesion and Migration in Neutrophils.
J Immunol.
2015; 195(12): 5770–9. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 45.
Picco A, Mund M, Ries J, et al.:
Visualizing the functional architecture of the endocytic machinery.
eLife.
2015; 4: e04535. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 46.
Han JJ, Kunde YA, Hong-Geller E, et al.:
Actin restructuring during Salmonella typhimurium infection investigated by confocal and super-resolution microscopy.
J Biomed Opt.
2014; 19(1): 16011. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 47.
Shroff H, Galbraith CG, Galbraith JA, et al.:
Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes.
Proc Natl Acad Sci U S A.
2007; 104(51): 20308–13. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 48.
Kner P, Chhun BB, Griffis ER, et al.:
Super-resolution video microscopy of live cells by structured illumination.
Nat Methods.
2009; 6(5): 339–42. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 49.
Vicidomini G, Hernández IC, d'Amora M, et al.:
Gated CW-STED microscopy: a versatile tool for biological nanometer scale investigation.
Methods.
2014; 66(2): 124–30. PubMed Abstract
| Publisher Full Text
- 50.
York AG, Parekh SH, Dalle Nogare D, et al.:
Resolution doubling in live, multicellular organisms via multifocal structured illumination microscopy.
Nat Methods.
2012; 9(7): 749–54. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 51.
Zhang X, Chen X, Zeng Z, et al.:
Development of a reversibly switchable fluorescent protein for super-resolution optical fluctuation imaging (SOFI).
ACS Nano.
2015; 9(3): 2659–67. PubMed Abstract
| Publisher Full Text
- 52.
Bálint Š, Verdeny Vilanova I, Sandoval Álvarez Á, et al.:
Correlative live-cell and superresolution microscopy reveals cargo transport dynamics at microtubule intersections.
Proc Natl Acad Sci U S A.
2013; 110(9): 3375–80. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 53.
Nakata T, Niwa S, Okada Y, et al.:
Preferential binding of a kinesin-1 motor to GTP-tubulin-rich microtubules underlies polarized vesicle transport.
J Cell Biol.
2011; 194(2): 245–55. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 54.
Xia P, Liu X, Wu B, et al.:
Superresolution imaging reveals structural features of EB1 in microtubule plus-end tracking.
Mol Biol Cell.
2014; 25(25): 4166–73. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 55.
Buss J, Coltharp C, Huang T, et al.:
In vivo organization of the FtsZ-ring by ZapA and ZapB revealed by quantitative super-resolution microscopy.
Mol Microbiol.
2013; 89(6): 1099–120. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 56.
Brodehl A, Hedde PN, Dieding M, et al.:
Dual color photoactivation localization microscopy of cardiomyopathy-associated desmin mutants.
J Biol Chem.
2012; 287(19): 16047–57. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 57.
Gustafsson MG:
Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy.
J Microsc.
2000; 198(Pt 2): 82–7. PubMed Abstract
| Publisher Full Text
- 58.
Gustafsson MG:
Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution.
Proc Natl Acad Sci U S A.
2005; 102(37): 13081–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 59.
Rego EH, Shao L, Macklin JJ, et al.:
Nonlinear structured-illumination microscopy with a photoswitchable protein reveals cellular structures at 50-nm resolution.
Proc Natl Acad Sci U S A.
2012; 109(3): E135–43. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 60.
Pielage J, Cheng L, Fetter RD, et al.:
A presynaptic giant ankyrin stabilizes the NMJ through regulation of presynaptic microtubules and transsynaptic cell adhesion.
Neuron.
2008; 58(2): 195–209. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 61.
Aslan JE, Phillips KG, Healy LD, et al.:
Histone deacetylase 6-mediated deacetylation of α-tubulin coordinates cytoskeletal and signaling events during platelet activation.
Am J Physiol Cell Physiol.
2013; 305(12): C1230–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 62.
Fu J, Glover DM:
Structured illumination of the interface between centriole and peri-centriolar material.
Open Biol.
2012; 2(8): 120104. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 63.
He R, Huang N, Bao Y, et al.:
LRRC45 is a centrosome linker component required for centrosome cohesion.
Cell Rep.
2013; 4(6): 1100–7. PubMed Abstract
| Publisher Full Text
- 64.
Chen CT, Kelly M, Leon JD, et al.:
Compartmentalized Toxoplasma EB1 bundles spindle microtubules to secure accurate chromosome segregation.
Mol Biol Cell.
2015; 26(25): 4562–76. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 65.
Fernandes I, Schöck F:
The nebulin repeat protein Lasp regulates I-band architecture and filament spacing in myofibrils.
J Cell Biol.
2014; 206(4): 559–72. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 66.
Granzier HL, Hutchinson KR, Tonino P, et al.:
Deleting titin's I-band/A-band junction reveals critical roles for titin in biomechanical sensing and cardiac function.
Proc Natl Acad Sci U S A.
2014; 111(40): 14589–94. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 67.
Beach JR, Shao L, Remmert K, et al.:
Nonmuscle myosin II isoforms coassemble in living cells.
Curr Biol.
2014; 24(10): 1160–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 68.
Brown AC, Oddos S, Dobbie IM, et al.:
Remodelling of cortical actin where lytic granules dock at natural killer cell immune synapses revealed by super-resolution microscopy.
PLoS Biol.
2011; 9(9): e1001152. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 69.
Santiago-Medina M, Gregus KA, Nichol RH, et al.:
Regulation of ECM degradation and axon guidance by growth cone invadosomes.
Development.
2015; 142(3): 486–96. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 70.
Whalen K, Reitzel AM, Hamdoun A:
Actin polymerization controls the activation of multidrug efflux at fertilization by translocation and fine-scale positioning of ABCB1 on microvilli.
Mol Biol Cell.
2012; 23(18): 3663–72. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 71.
Shimi T, Kittisopikul M, Tran J, et al.:
Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy.
Mol Biol Cell.
2015; 26(22): 4075–86. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 72.
Stahley SN, Warren MF, Feldman RJ, et al.:
Super-Resolution Microscopy Reveals Altered Desmosomal Protein Organization in Tissue from Patients with Pemphigus Vulgaris.
J Invest Dermatol.
2016; 136(1): 59–66. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 73.
Hu S, Tee YH, Kabla A, et al.:
Structured illumination microscopy reveals focal adhesions are composed of linear subunits.
Cytoskeleton (Hoboken).
2015; 72(5): 235–45. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 74.
Case LB, Baird MA, Shtengel G, et al.:
Molecular mechanism of vinculin activation and nanoscale spatial organization in focal adhesions.
Nat Cell Biol.
2015; 17(7): 880–92. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 75.
Kanchanawong P, Shtengel G, Pasapera AM, et al.:
Nanoscale architecture of integrin-based cell adhesions.
Nature.
2010; 468(7323): 580–4. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 76.
Liu J, Wang Y, Goh WI, et al.:
Talin determines the nanoscale architecture of focal adhesions.
Proc Natl Acad Sci U S A.
2015; 112(35): E4864–73. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 77.
Guillaume E, Comunale F, Do Khoa N, et al.:
Flotillin microdomains stabilize cadherins at cell-cell junctions.
J Cell Sci.
2013; 126(Pt 22): 5293–304. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 78.
Oldenburg J, van der Krogt G, Twiss F, et al.:
VASP, zyxin and TES are tension-dependent members of Focal Adherens Junctions independent of the α-catenin-vinculin module.
Sci Rep.
2015; 5: 17225. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 79.
Westin L, Reuss M, Lindskog M, et al.:
Nanoscopic spine localization of Norbin, an mGluR5 accessory protein.
BMC Neurosci.
2014; 15: 45. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 80.
Sonnen KF, Schermelleh L, Leonhardt H, et al.:
3D-structured illumination microscopy provides novel insight into architecture of human centrosomes.
Biol Open.
2012; 1(10): 965–76. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 81.
Wynne DJ, Funabiki H:
Kinetochore function is controlled by a phospho-dependent coexpansion of inner and outer components.
J Cell Biol.
2015; 210(6): 899–916. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 82.
Duleh SN, Welch MD:
WASH and the Arp2/3 complex regulate endosome shape and trafficking.
Cytoskeleton (Hoboken).
2010; 67(3): 193–206. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 83.
Hell SW:
Far-field optical nanoscopy.
Science.
2007; 316(5828): 1153–8. PubMed Abstract
| Publisher Full Text
- 84.
Klar TA, Hell SW:
Subdiffraction resolution in far-field fluorescence microscopy.
Opt Lett.
1999; 24(14): 954–6. PubMed Abstract
| Publisher Full Text
- 85.
Hofmann M, Eggeling C, Jakobs S, et al.:
Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins.
Proc Natl Acad Sci U S A.
2005; 102(49): 17565–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 86.
Hein B, Willig KI, Hell SW:
Stimulated emission depletion (STED) nanoscopy of a fluorescent protein-labeled organelle inside a living cell.
Proc Natl Acad Sci U S A.
2008; 105(38): 14271–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 87.
Westphal V, Lauterbach MA, Di Nicola A, et al.:
Dynamic far-field fluorescence nanoscopy.
New J Phys.
2007; 9: 435. Publisher Full Text
- 88.
Westphal V, Rizzoli SO, Lauterbach MA, et al.:
Video-rate far-field optical nanoscopy dissects synaptic vesicle movement.
Science.
2008; 320(5873): 246–9. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 89.
D'Este E, Kamin D, Göttfert F, et al.:
STED nanoscopy reveals the ubiquity of subcortical cytoskeleton periodicity in living neurons.
Cell Rep.
2015; 10(8): 1246–51. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 90.
Urban NT, Willig KI, Hell SW, et al.:
STED nanoscopy of actin dynamics in synapses deep inside living brain slices.
Biophys J.
2011; 101(5): 1277–84. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 91.
Reimold C, Defeu Soufo HJ, Dempwolff F, et al.:
Motion of variable-length MreB filaments at the bacterial cell membrane influences cell morphology.
Mol Biol Cell.
2013; 24(15): 2340–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 92.
Rak GD, Mace EM, Banerjee PP, et al.:
Natural killer cell lytic granule secretion occurs through a pervasive actin network at the immune synapse.
PLoS Biol.
2011; 9(9): e1001151. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 93.
Tamarit B, Bugault F, Pillet AH, et al.:
Membrane microdomains and cytoskeleton organization shape and regulate the IL-7 receptor signalosome in human CD4 T-cells.
J Biol Chem.
2013; 288(12): 8691–701. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 94.
Shutova MS, Spessott WA, Giraudo CG, et al.:
Endogenous species of mammalian nonmuscle myosin IIA and IIB include activated monomers and heteropolymers.
Curr Biol.
2014; 24(17): 1958–68. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 95.
Ashour DJ, Pelka B, Jaaks P, et al.:
The catalytic domain of inositol-1,4,5-trisphosphate 3-kinase-a contributes to ITPKA-induced modulation of F-actin.
Cytoskeleton (Hoboken).
2015; 72(2): 93–100. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 96.
Gad AK, Rönnlund D, Spaar A, et al.:
Rho GTPases link cellular contractile force to the density and distribution of nanoscale adhesions.
FASEB J.
2012; 26(6): 2374–82. PubMed Abstract
| Publisher Full Text
- 97.
Yang TT, Hampilos PJ, Nathwani B, et al.:
Superresolution STED microscopy reveals differential localization in primary cilia.
Cytoskeleton (Hoboken).
2013; 70(1): 54–65. PubMed Abstract
| Publisher Full Text
- 98.
Oddoux S, Zaal KJ, Tate V, et al.:
Microtubules that form the stationary lattice of muscle fibers are dynamic and nucleated at Golgi elements.
J Cell Biol.
2013; 203(2): 205–13. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 99.
Rothmeier E, Pfaffinger G, Hoffmann C, et al.:
Activation of Ran GTPase by a Legionella effector promotes microtubule polymerization, pathogen vacuole motility and infection.
PLoS Pathog.
2013; 9(9): e1003598. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 100.
Rathje LS, Nordgren N, Pettersson T, et al.:
Oncogenes induce a vimentin filament collapse mediated by HDAC6 that is linked to cell stiffness.
Proc Natl Acad Sci U S A.
2014; 111(4): 1515–20. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 101.
Rönnlund D, Gad AK, Blom H, et al.:
Spatial organization of proteins in metastasizing cells.
Cytometry A.
2013; 83(9): 855–65. PubMed Abstract
| Publisher Full Text
- 102.
Kayser J, Grabmayr H, Harasim M, et al.:
Assembly kinetics determine the structure of keratin networks.
Soft Matter.
2012; 8: 8873–8879. Publisher Full Text
- 103.
Depreux FF, Puckelwartz MJ, Augustynowicz A, et al.:
Disruption of the lamin A and matrin-3 interaction by myopathic LMNA mutations.
Hum Mol Genet.
2015; 24(15): 4284–95. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 104.
Bhattacharya R, Gonzalez AM, Debiase PJ, et al.:
Recruitment of vimentin to the cell surface by beta3 integrin and plectin mediates adhesion strength.
J Cell Sci.
2009; 122(Pt 9): 1390–400. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 105.
Gonzales M, Weksler B, Tsuruta D, et al.:
Structure and function of a vimentin-associated matrix adhesion in endothelial cells.
Mol Biol Cell.
2001; 12(1): 85–100. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 106.
Frigault MM, Lacoste J, Swift JL, et al.:
Live-cell microscopy - tips and tools.
J Cell Sci.
2009; 122( Pt 6): 753–67. PubMed Abstract
| Publisher Full Text
- 107.
Schneckenburger H, Weber P, Wagner M, et al.:
Light exposure and cell viability in fluorescence microscopy.
J Microsc.
2012; 245(3): 311–8. PubMed Abstract
| Publisher Full Text
- 108.
Wäldchen S, Lehmann J, Klein T, et al.:
Light-induced cell damage in live-cell super-resolution microscopy.
Sci Rep.
2015; 5: 15348. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 109.
Vigers GP, Coue M, McIntosh JR:
Fluorescent microtubules break up under illumination.
J Cell Biol.
1988; 107(3): 1011–24. PubMed Abstract
| Free Full Text
- 110.
Fernández-Martín L, Marcos-Ramiro B, Bigarella CL, et al.:
Crosstalk between reticular adherens junctions and platelet endothelial cell adhesion molecule-1 regulates endothelial barrier function.
Arterioscler Thromb Vasc Biol.
2012; 32(8): e90–102. PubMed Abstract
| Publisher Full Text
- 111.
Guizetti J, Schermelleh L, Mäntler J, et al.:
Cortical constriction during abscission involves helices of ESCRT-III-dependent filaments.
Science.
2011; 331(6024): 1616–20. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 112.
Luo W, Yu CH, Lieu ZZ, et al.:
Analysis of the local organization and dynamics of cellular actin networks.
J Cell Biol.
2013; 202(7): 1057–73. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 113.
Versaevel M, Braquenier JB, Riaz M, et al.:
Super-resolution microscopy reveals LINC complex recruitment at nuclear indentation sites.
Sci Rep.
2014; 4: 7362. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 114.
Komis G, Mistrik M, Samajová O, et al.:
Dynamics and organization of cortical microtubules as revealed by superresolution structured illumination microscopy.
Plant Physiol.
2014; 165(1): 129–48. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 115.
Zobel T, Bogdan S:
A high resolution view of the fly actin cytoskeleton lacking a functional WAVE complex.
J Microsc.
2013; 251(3): 224–31. PubMed Abstract
| Publisher Full Text
- 116.
Burnette DT, Shao L, Ott C, et al.:
A contractile and counterbalancing adhesion system controls the 3D shape of crawling cells.
J Cell Biol.
2014; 205(1): 83–96. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 117.
Indra I, Hong S, Troyanovsky R, et al.:
The adherens junction: a mosaic of cadherin and nectin clusters bundled by actin filaments.
J Invest Dermatol.
2013; 133(11): 2546–54. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 118.
Hookway C, Ding L, Davidson MW, et al.:
Microtubule-dependent transport and dynamics of vimentin intermediate filaments.
Mol Biol Cell.
2015; 26(9): 1675–86. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 119.
Shroff H, Galbraith CG, Galbraith JA, et al.:
Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics.
Nat Methods.
2008; 5(5): 417–23. PubMed Abstract
| Publisher Full Text
- 120.
Wilmes S, Staufenbiel M, Lisse D, et al.:
Triple-color super-resolution imaging of live cells: resolving submicroscopic receptor organization in the plasma membrane.
Angew Chem Int Ed Engl.
2012; 51(20): 4868–71. PubMed Abstract
| Publisher Full Text
- 121.
Mudrakola HV, Zhang K, Cui B:
Optically resolving individual microtubules in live axons.
Structure.
2009; 17(11): 1433–41. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 122.
Chazeau A, Mehidi A, Nair D, et al.:
Nanoscale segregation of actin nucleation and elongation factors determines dendritic spine protrusion.
EMBO J.
2014; 33(23): 2745–64. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 123.
Rossier O, Octeau V, Sibarita JB, et al.:
Integrins β1 and β3 exhibit distinct dynamic nanoscale organizations inside focal adhesions.
Nat Cell Biol.
2012; 14(10): 1057–67. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 124.
Tatavarty V, Das S, Yu J:
Polarization of actin cytoskeleton is reduced in dendritic protrusions during early spine development in hippocampal neuron.
Mol Biol Cell.
2012; 23(16): 3167–77. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 125.
Tatavarty V, Kim EJ, Rodionov V, et al.:
Investigating sub-spine actin dynamics in rat hippocampal neurons with super-resolution optical imaging.
PLoS One.
2009; 4(11): e7724. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 126.
Ding JB, Takasaki KT, Sabatini BL:
Supraresolution imaging in brain slices using stimulated-emission depletion two-photon laser scanning microscopy.
Neuron.
2009; 63(4): 429–37. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 127.
Li Q, Wu SS, Chou KC:
Subdiffraction-limit two-photon fluorescence microscopy for GFP-tagged cell imaging.
Biophys J.
2009; 97(12): 3224–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 128.
Moneron G, Hell SW:
Two-photon excitation STED microscopy.
Opt Express.
2009; 17(17): 14567–73. PubMed Abstract
| Publisher Full Text
- 129.
Gould TJ, Myers JR, Bewersdorf J:
Total internal reflection STED microscopy.
Opt Express.
2011; 19(14): 13351–7. PubMed Abstract
| Publisher Full Text
- 130.
Gustafsson MG, Shao L, Carlton PM, et al.:
Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination.
Biophys J.
2008; 94(12): 4957–70. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 131.
Shao L, Kner P, Rego EH, et al.:
Super-resolution 3D microscopy of live whole cells using structured illumination.
Nat Methods.
2011; 8(12): 1044–6. PubMed Abstract
| Publisher Full Text
- 132.
Strauss MP, Liew AT, Turnbull L, et al.:
3D-SIM super resolution microscopy reveals a bead-like arrangement for FtsZ and the division machinery: implications for triggering cytokinesis.
PLoS Biol.
2012; 10(9): e1001389. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 133.
Huang B, Jones SA, Brandenburg B, et al.:
Whole-cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution.
Nat Methods.
2008; 5(12): 1047–52. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 134.
Xu K, Babcock HP, Zhuang X:
Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton.
Nat Methods.
2012; 9(2): 185–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 135.
Harke B, Ullal CK, Keller J, et al.:
Three-dimensional nanoscopy of colloidal crystals.
Nano Lett.
2008; 8(5): 1309–13. PubMed Abstract
| Publisher Full Text
- 136.
Böhm U, Hell SW, Schmidt R:
4Pi-RESOLFT nanoscopy.
Nat Commun.
2016; 7: 10504. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 137.
Shelden E, Knecht DA:
Mutants lacking myosin II cannot resist forces generated during multicellular morphogenesis.
J Cell Sci.
1995; 108(Pt 3): 1105–15. PubMed Abstract
- 138.
Wu PH, Giri A, Sun SX, et al.:
Three-dimensional cell migration does not follow a random walk.
Proc Natl Acad Sci U S A.
2014; 111(11): 3949–54. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 139.
Cella Zanacchi F, Lavagnino Z, Perrone Donnorso M, et al.:
Live-cell 3D super-resolution imaging in thick biological samples.
Nat Methods.
2011; 8(12): 1047–9. PubMed Abstract
| Publisher Full Text
- 140.
Gao L, Shao L, Higgins CD, et al.:
Noninvasive imaging beyond the diffraction limit of 3D dynamics in thickly fluorescent specimens.
Cell.
2012; 151(6): 1370–85. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 141.
Winter PW, York AG, Nogare DD, et al.:
Two-photon instant structured illumination microscopy improves the depth penetration of super-resolution imaging in thick scattering samples.
Optica.
2014; 1(3): 181–91. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 142.
Bartesaghi A, Merk A, Banerjee S, et al.:
2.2 Å resolution cryo-EM structure of β-galactosidase in complex with a cell-permeant inhibitor.
Science.
2015; 348(6239): 1147–51. PubMed Abstract
| Publisher Full Text
- 143.
Phillipp F:
Advances in High-Resolution Transmission Electron Microscopy.
Materials Transactions JIM.
1998; 39(9): 888–902. Publisher Full Text
Comments on this article Comments (0)