Keywords
Genomics, Intervals, BEDtools, reproducibility, R, RStudio,
This article is included in the RPackage gateway.
Genomics, Intervals, BEDtools, reproducibility, R, RStudio,
A routine bioinformatic task is the analysis of the relationships between sets of genomic intervals, including the identification of DNA variants within protein coding regions, annotation of regions enriched for nucleic acid binding proteins, and computation of read density within a set of exons. Command-line tools for interval analysis such as BEDtools1 and BEDOPS2 enable analyses of genome-wide datasets and are key components of analysis pipelines. Analyses with these tools commonly combine processing intervals on the command-line with visualization and statistical analysis in R. However, the need to master both the command-line and R hinders exploratory data analysis, and the development of reproducible research workflows built in the RMarkdown framework.
Existing R packages developed for interval analysis include IRanges3, bedr4, and GenometriCorr5. IRanges is a Bioconductor package that provides interval classes and methods to perform interval arithmetic, and is used by many Bioconductor packages. bedr is a CRAN-distributed package that provides wrapper R functions to call the BEDtools, BEDOPS, and tabix command-line utilities, providing out-of-memory support for interval analysis. Finally, GenometriCorr provides a set of statistical tests to determine the relationships between interval sets using IRanges data structures. These packages provide functionality for processing and statistical inference of interval data, however they require a detailed understanding of S4 classes (IRanges) or the installation of external command-line dependencies (bedr). Additionally, these packages do not easily integrate with the recent advances provided by the popular tidyverse suite of data processing and visualization tools (e.g. dplyr, purrr, broom and ggplot2)6. We therefore sought to develop a flexible R package for genomic interval arithmetic built to incorporate new R programming, visualization, and interactivity features.
valr is an R package that makes extensive use of dplyr, a flexible and high-performance framework for data manipulation in R7. Additionally, compute intensive functions in valr are written in C++ using Rcpp to enable fluid interactive analysis of large datasets8. Interval intersections and related operations use an interval tree algorithm to efficiently search for overlapping intervals9. BED files are imported and handled in R as data_frame objects, requiring minimal pre or post-processing to integrate with additional R packages or command-line tools.
valr is distributed as part of the CRAN R package repository and is compatible with Mac OS X, Windows, and major Linux operating systems. Package dependencies and system requirements are documented in the valr CRAN repository.
To demonstrate the functionality and utility of valr, we present a basic tutorial for using valr and additional common use cases for genomic interval analysis.
Input data. valr provides a set of functions to read BED, BEDgraph, and VCF formats into R as convenient tibble (tbl) data_frame objects. All tbls have chrom, start, and end columns, and tbls from multi-column formats have additional pre-determined column names. Standards methods for importing data (e.g. read.table, readr::read_tsv) are also supported provided the constructed dataframes contain the requisite column names (chrom, start, end). Additionally, valr supports connections to remote databases to access the UCSC and Ensembl databases via the db_ucsc and db_ensembl functions.
library(valr) # function to retrieve path to example data bed_filepath <- valr_example("3fields.bed.gz") read_bed(bed_filepath) #> # A tibble: 10 x 3 #> chrom start end #> <chr> <int> <int> #> 1 chr1 11873 14409 #> 2 chr1 14361 19759 #> 3 chr1 14406 29370 #> 4 chr1 34610 36081 #> 5 chr1 69090 70008 #> 6 chr1 134772 140566 #> 7 chr1 321083 321115 #> 8 chr1 321145 321207 #> 9 chr1 322036 326938 #> 10 chr1 327545 328439
#using URL read_bed("https://github.com/rnabioco/valr/raw/master/inst/extdata/3fields.bed.gz") #> # A tibble: 10 x 3 #> chrom start end #> <chr> <int> <int> #> 1 chr1 11873 14409 #> 2 chr1 14361 19759 #> 3 chr1 14406 29370 #> 4 chr1 34610 36081 #> 5 chr1 69090 70008 #> 6 chr1 134772 140566 #> 7 chr1 321083 321115 #> 8 chr1 321145 321207 #> 9 chr1 322036 326938 #> 10 chr1 327545 328439
Example of combining valr tools. The functions in valr have similar names to their BEDtools counterparts, and so will be familiar to users of the BEDtools suite. Also, similar to pybedtools10, a python wrapper for BEDtools, valr has a terse syntax. For example, shown below is a demonstration of how to find all intergenic SNPs within 1 kilobase of genes using valr. The BED files used in the following examples are described in the Data Availability section.
library(dplyr) snps <- read_bed(valr_example("hg19.snps147.chr22.bed.gz"), n_fields = 6) genes <- read_bed(valr_example("genes.hg19.chr22.bed.gz"), n_fields = 6) # find snps in intergenic regions intergenic <- bed_subtract(snps, genes) # distance from intergenic snps to nearest gene nearby <- bed_closest(intergenic, genes) nearby %>% select(starts_with("name"), .overlap, .dist) %>% filter(abs(.dist) < 1000) #> # A tibble: 285 x 4 #> name.x name.y .overlap .dist #> <chr> <chr> <int> <int> #> 1 rs2261631 P704P 0 -267 #> 2 rs570770556 POTEH 0 -912 #> 3 rs538163832 POTEH 0 -952 #> 4 rs9606135 TPTEP1 0 -421 #> 5 rs11912392 ANKRD62P1-PARP4P3 0 104 #> 6 rs8136454 BC038197 0 355 #> 7 rs5992556 XKR3 0 -455 #> 8 rs114101676 GAB4 0 473 #> 9 rs62236167 CECR7 0 261 #> 10 rs5747023 CECR1 0 -386 #> # ... with 275 more rows
Visual documentation. By conducting interval arithmetic entirely in R, valr is also an effective teaching tool for introducing interval analysis to early-stage analysts without requiring familiarity with both command-line tools and R. To aid in demonstrating the interval operations available in valr, we developed the bed_glyph() tool which produces plots demonstrating the input and output of operations in valr in a manner similar to those found in the BEDtools documentation. Shown below is the code required to produce glyphs displaying the results of intersecting x and y intervals with bed_intersect(), and the result of merging x intervals with bed_merge() (Figure 1).
x <- tibble::tribble( ~chrom, ~start, ~end, "chr1", 25, 50, "chr1", 100, 125 ) y <- tibble::tribble( ~chrom, ~start, ~end, "chr1", 30, 75 ) bed_glyph(bed_intersect(x, y))
And this glyph illustrates bed_merge():
x <- tibble::tribble( ~chrom, ~start, ~end, "chr1", 1, 50, "chr1", 10, 75, "chr1", 100, 120 ) bed_glyph(bed_merge(x))

Grouping data. The group_by function in dplyr can be used to execute functions on subsets of single and multiple data_frames. Functions in valr leverage grouping to enable a variety of comparisons. For example, intervals can be grouped by strand to perform comparisons among intervals on the same strand.
x <- tibble::tribble( ~chrom, ~start, ~end, ~strand, "chr1", 1, 100, "+", "chr1", 50, 150, "+", "chr2", 100, 200, "-" ) y <- tibble::tribble( ~chrom, ~start, ~end, ~strand, "chr1", 50, 125, "+", "chr1", 50, 150, "-", "chr2", 50, 150, "+" ) # intersect tbls by strand x <- group_by(x, strand) y <- group_by(y, strand) bed_intersect(x, y) #> # A tibble: 2 x 8 #> chrom start.x end.x strand.x start.y end.y strand.y .overlap #> <chr> <dbl> <dbl> <chr> <dbl> <dbl> <chr> <int> #> 1 chr1 1 100 + 50 125 + 50 #> 2 chr1 50 150 + 50 125 + 75
Comparisons between intervals on opposite strands are done using the flip_strands() function:
x <- group_by(x, strand) y <- flip_strands(y) y <- group_by(y, strand) bed_intersect(x, y) #> # A tibble: 3 x 8 #> chrom start.x end.x strand.x start.y end.y strand.y .overlap #> <chr> <dbl> <dbl> <chr> <dbl> <dbl> <chr> <int> #> 1 chr2 100 200 - 50 150 - 50 #> 2 chr1 1 100 + 50 150 + 50 #> 3 chr1 50 150 + 50 150 + 100
Both single set (e.g. bed_merge()) and multi set operations will respect groupings in the input intervals.
Column specification. Columns in BEDtools are referred to by position:
# calculate the mean of column 6 for intervals in ‘b‘ that overlap with ‘a‘ bedtools map -a a.bed -b b.bed -c 6 -o mean
In valr, columns are referred to by name and can be used in multiple name/value expressions for summaries.
# calculate the mean and variance for a ‘value‘ column bed_map(a, b, .mean = mean(value), .var = var(value)) # report concatenated and max values for merged intervals bed_merge(a, .concat = concat(value), .max = max(value))
API. The major functions available in valr are shown in Table 1.
This demonstration illustrates how to use valr tools to perform a “meta-analysis” of signals relative to genomic features. Here we analyze the distribution of histone marks surrounding transcription start sites, using H3K4Me3 Chip-Seq data from the ENCODE project.
First we load packages and relevant data.
bedfile <- valr_example("genes.hg19.chr22.bed.gz") genomefile <- valr_example("hg19.chrom.sizes.gz") bgfile <- valr_example("hela.h3k4.chip.bg.gz") genes <- read_bed(bedfile, n_fields = 6) genome <- read_genome(genomefile) y <- read_bedgraph(bgfile)
Then, we generate 1 bp intervals to represent transcription start sites (TSSs). We focus on + strand genes, but - genes are easily accommodated by filtering them and using bed_makewindows() with reversed window numbers.
# generate 1 bp TSS intervals, "+" strand only tss <- genes %>% filter(strand == "+") %>% mutate(end = start + 1) # 1000 bp up and downstream region_size <- 1000 # 50 bp windows win_size <- 50 # add slop to the TSS, break into windows and add a group x <- tss %>% bed_slop(genome, both = region_size) %>% bed_makewindows(genome, win_size) x #> # A tibble: 13,530 x 7 #> chrom start end name score strand .win_id #> <chr> <int> <int> <chr> <chr> <chr> <int> #> 1 chr22 16161065 16161115 LINC00516 3 + 1 #> 2 chr22 16161115 16161165 LINC00516 3 + 2 #> 3 chr22 16161165 16161215 LINC00516 3 + 3 #> 4 chr22 16161215 16161265 LINC00516 3 + 4 #> 5 chr22 16161265 16161315 LINC00516 3 + 5 #> 6 chr22 16161315 16161365 LINC00516 3 + 6 #> 7 chr22 16161365 16161415 LINC00516 3 + 7 #> 8 chr22 16161415 16161465 LINC00516 3 + 8 #> 9 chr22 16161465 16161515 LINC00516 3 + 9 #> 10 chr22 16161515 16161565 LINC00516 3 + 10 #> # ... with 13,520 more rows
Now we use the .win_id group with bed_map() to calculate a sum by mapping y signals onto the intervals in x. These data are regrouped by .win_id and a summary with mean and sd values is calculated.
# map signals to TSS regions and calculate summary statistics. res <- bed_map(x, y, win_sum = sum(value, na.rm = TRUE)) %>% group_by(.win_id) %>% summarize(win_mean = mean(win_sum, na.rm = TRUE), win_sd = sd(win_sum, na.rm = TRUE)) res #> # A tibble: 41 x 3 #> .win_id win_mean win_sd #> <int> <dbl> <dbl> #> 1 1 100.8974 85.83423 #> 2 2 110.6829 81.13521 #> 3 3 122.9070 99.09635 #> 4 4 116.2800 96.30098 #> 5 5 116.3500 102.33773 #> 6 6 124.9048 95.08887 #> 7 7 122.9437 94.39792 #> 8 8 127.5946 91.47407 #> 9 9 130.2051 95.71309 #> 10 10 130.1220 88.82809 #> # ... with 31 more rows
Finally, these summary statistics are used to construct a plot that illustrates histone density surrounding TSSs (Figure 2).
library(ggplot2) x_labels <- seq(-region_size, region_size, by = win_size ∗ 5) x_breaks <- seq(1, 41, by = 5) sd_limits <- aes(ymax = win_mean + win_sd, ymin = win_mean - win_sd) p <- ggplot(res, aes(x = .win_id, y = win_mean)) + geom_point(size = 0.25) + geom_pointrange(sd_limits, size = 0.1) + scale_x_continuous(labels = x_labels, breaks = x_breaks) + xlab("Position (bp from TSS)") + ylab("Signal") + theme_classic()

(A) Summarized coverage of human H3K4Me3 Chip-Seq coverage across positive strand transcription start sites on chromosome 22. Data presented +/- SD.
Estimates of significance for interval overlaps can be obtained by combining bed_shuffle(), bed_random() and the sample_ functions from dplyr with interval statistics in valr.
Here, we examine the extent of overlap of repeat classes (repeatmasker track obtained from the UCSC genome browser) with exons in the human genome (hg19 build, on chr22 only, for simplicity) using the jaccard similarity index. bed_jaccard() implements the jaccard test to examine the similarity between two sets of genomic intervals. Using bed_shuffle() and replicate() we generate a data_frame containing 100 sets of randomly selected intervals then calculate the jaccard index for each set against the repeat intervals to generate a null-distribution of jaccard scores. Finally, an empirical p-value is calculated from the null-distribution.
library(tidyverse) repeats <- read_bed(valr_example("hg19.rmsk.chr22.bed.gz"), n_fields = 6) genome <- read_genome(valr_example("hg19.chrom.sizes.gz")) genes <- read_bed12(valr_example("hg19.refGene.chr22.bed.gz")) # convert bed12 to bed with exons exons <- bed12_to_exons(genes) # function to repeat interval shuffling shuffle_intervals <- function(n, .data, genome) { replicate(n, bed_shuffle(.data, genome), simplify = FALSE) %>% bind_rows(.id = "rep") %>% group_by(rep) %>% nest() } nreps <- 100
shuffled <- shuffle_intervals(n = nreps, repeats, genome) %>% mutate(jaccard = data %>% map(bed_jaccard, repeats) %>% map_dbl("jaccard")) shuffled #> # A tibble: 100 x 3 #> rep data jaccard #> <chr> <list> <dbl> #> 1 1 <tibble [10,000 x 6]> 0.0003388967 #> 2 2 <tibble [10,000 x 6]> 0.0004965988 #> 3 3 <tibble [10,000 x 6]> 0.0002974843 #> 4 4 <tibble [10,000 x 6]> 0.0006899870 #> 5 5 <tibble [10,000 x 6]> 0.0004678412 #> 6 6 <tibble [10,000 x 6]> 0.0001726937 #> 7 7 <tibble [10,000 x 6]> 0.0004694941 #> 8 8 <tibble [10,000 x 6]> 0.0004660410 #> 9 9 <tibble [10,000 x 6]> 0.0006846643 #> 10 10 <tibble [10,000 x 6]> 0.0002143829 #> # ... with 90 more rows obs <- bed_jaccard(repeats, exons) obs #> # A tibble: 1 x 4 #> len_i len_u jaccard n #> <dbl> <dbl> <dbl> <dbl> #> 1 112123 4132109 0.02789139 805 pvalue <- sum(shuffled$jaccard >= obs$jaccard) + 1 /(nreps + 1) pvalue #> [1] 0.00990099
In order to ensure that valr performs fast enough to enable interactive analysis, key functionality is implemented in C++. To test the speed of major valr functions we generated two data_frames containing 1 million randomly selected 1 kilobase intervals derived from the human genome (hg19). Most of the major valr functions complete execution in less than 1 second, demonstrating that valr can process large interval datasets efficiently (Figure 3A).
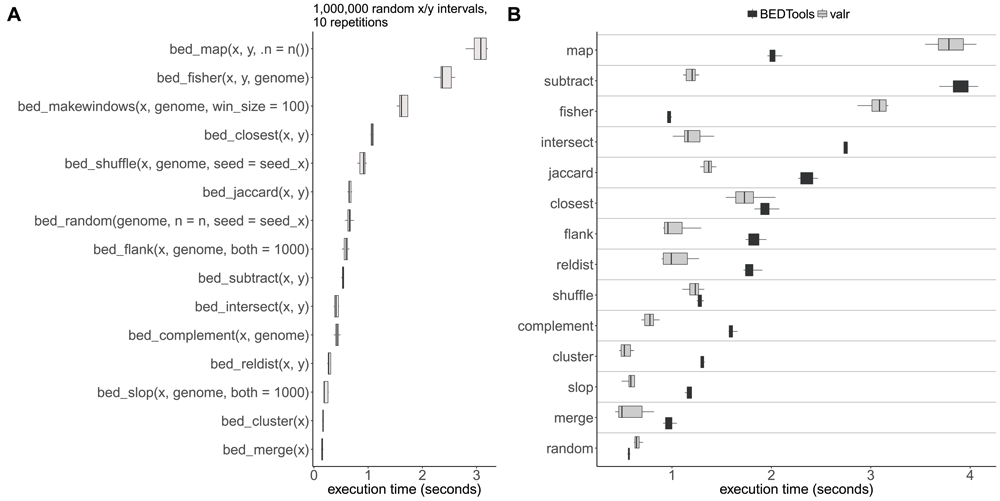
(A) Timings were calculated by performing 10 repetitions of indicated functions on data frames preloaded in R containing 1 million random 1 kilobase x/y intervals generated using bed_random(). (B) Timings for executing functions in BEDtools v2.25.0 or equivalent functions in valr using the same interval sets as in (A) written to files. All BEDtools function outputs were written to /dev/null/, and were timed using GNU time. Timings for valr functions in (B) include times for reading files using read_bed() functions and were timed using the microbenchmark package.
We also benchmarked major valr functions against corresponding commands in BEDtools. valr operates on data_frames already loaded into RAM, whereas BEDtools performs file-reading, processing, and writing. To compare valr against BEDtools we generated two BED files containing 1 million randomly selected 1 kilobase intervals derived from the human genome (hg19). For valr functions, we timed reading the table into R (e.g. with read_bed()) and performing the respective function. For BEDtools commands we timed executing the command with the output written to /dev/null. valr functions performed similarly or faster than BEDtools commands, with the exception of bed_map and bed_fisher (Figure 3B).
Command-line tools like BEDtools and bedops can be incorporated into reproducible workflows (e.g., with snakemake11), but it is cumbersome to transition from command-line tools to exploratory analysis and plotting software. RMarkdown documents are plain text files, amenable to version control, which provide an interface to generate feature rich PDF and HTML reports that combine text, executable code, and figures in a single document. valr can be used in RMarkdown documents to provide rapid documentation of exploratory data analyses and generate reproducible work-flows for data processing. Moreover, new features in RStudio, such as notebook viewing, and multiple language support enable similar functionality to another popular notebook platform jupyter notebooks.
Additionally, valr seamlessly integrates into R shiny12 applications allowing for complex interactive visualizations relating to genomic interval analyses. We have developed a shiny application (available on Gitub) that explores ChiP-Seq signal density surrounding transcription start sites and demonstrates the ease of implementing valr to power dynamic visualizations.
valr provides a flexible framework for interval arithmetic in R/Rstudio. valr functions are written with a simple and terse syntax that promotes flexible interactive analysis. Additionally by providing an easy-to-use interface for interval arithmetic in R, valr is also a useful teaching tool to introduce the analyses necessary to investigate correlations between genomic intervals, without requiring familiarity with the command-line. We envision that valr will help researchers quickly and reproducibly analyze genome interval datasets.
The valr package includes external datasets stored in the inst/extdata/ directory that were used in this manuscript. These datasets were obtained from the ENCODE Project13 or the UCSC genome browser14. BED files were generated by converting the UCSC tables into BED format. BED and BEDgraph data was only kept from chromosome 22, and was subsampled to produce file sizes suitable for submission to the CRAN repository. The original raw data is available from the following sources:
hela.h3k4.chip.bg.gz SRA record: SRR227441, ENCODE identifier: ENCSR000AOF
hg19.refGene.chr22.bed.gz ftp://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/refGene.txt.gz
hg19.rmsk.chr22.bed.gz ftp://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/rmsk.txt.gz
hg19.chrom.sizes.gz ftp://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/chromInfo.txt.gz
genes.hg19.chr22.bed.gz ftp://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/refGene.txt.gz
hg19.snps147.chr22.bed.gz ftp://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/snp147.txt.gz
valr can be installed via CRAN using install.packages("valr").
valr is maintained at http://github.com/rnabioco/valr.
Latest valr source code is available at http://github.com/rnabioco/valr.
The latest stable version of source code is at: https://github.com/rnabioco/valr/archive/v0.3.0.tar.gz
Archived source code at the time of publication: http://doi.org/10.5281/zenodo.81540315
License: MIT license.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)