1Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, 1250 Moursund St, Houston, TX, 77030, USA 2Department of Molecular and Human Genetics, Baylor College of Medicine, One Baylor Plaza, Houston, TX, 77030, USA 3Parkinson's Disease Center and Movement Disorders Clinic, Department of Neurology, Baylor College of Medicine, 7200 Cambridge, Houston, TX, 77030-4202, USA 4Department of Neuroscience, Baylor College of Medicine, One Baylor Plaza, Houston, TX, 77030, USA
Maxime W.C. Rousseaux
Roles:
Writing – Original Draft Preparation,
Writing – Review & Editing
Joshua M. Shulman
Roles:
Writing – Original Draft Preparation,
Writing – Review & Editing
Joseph Jankovic
Roles:
Writing – Original Draft Preparation,
Writing – Review & Editing
OPEN PEER REVIEW
REVIEWER STATUS
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disorder after Alzheimer’s disease, affecting over 10 million individuals worldwide. While numerous effective symptomatic treatments are currently available, no curative or disease-modifying therapies exist. An integrated, comprehensive understanding of PD pathogenic mechanisms will likely address this unmet clinical need. Here, we highlight recent progress in PD research with an emphasis on promising translational findings, including (i) advances in our understanding of disease susceptibility, (ii) improved knowledge of cellular dysfunction, and (iii) insights into mechanisms of spread and propagation of PD pathology. We emphasize connections between these previously disparate strands of PD research and the development of an emerging systems-level understanding that will enable the next generation of PD therapeutics.
Corresponding author:
Joseph Jankovic
Competing interests:
Maxime W.C. Rousseaux and Joshua M. Shulman declare that they have no disclosures. Joseph Jankovic has received research or training grants and/or has served as a consultant for the Michael J. Fox Foundation for Parkinson’s Research, Prothena Biosciences Inc, and Teva Pharmaceutical Industries Ltd.
Grant information:
Maxime W.C. Rousseaux is supported in part by Grant No. PF-JFA-1762 from the Parkinson’s Disease Foundation. Joshua M. Shulman is supported by the Huffington Foundation, the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital, and a Career Award for Medical Scientists from the Burroughs Wellcome Fund. Joseph Jankovic has received research and/or educational support from the Michael J. Fox Foundation for Parkinson’s Research, Parkinson’s Foundation, the Parkinson Study Group, Prothena Biosciences Inc, and Teva Pharmaceutical Industries Ltd.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Parkinson’s disease (PD) is the most common movement disorder, affecting 2–3% of individuals over the age of 651. It is clinically characterized by a core set of motor manifestations, including tremor, slow movement (bradykinesia), increased muscle tone (rigidity), and gait and postural impairment as well as a variety of other motor and non-motor features, including cognitive impairment, depression, pain and other sensory symptoms, autonomic dysfunction, and others2. PD is characterized pathologically by the loss of predominantly dopaminergic neurons, associated with intracellular, insoluble α-synuclein (α-Syn) aggregates, largely localized to cytoplasmic inclusions termed Lewy bodies and within neuronal processes termed Lewy neurites. Whereas current treatments can ameliorate the cardinal motor symptoms, no disease-modifying therapies exist3. A large body of research has therefore focused on understanding the biological mechanisms that underlie disease onset and progression, with the goal of developing effective pathogenesis-targeted, disease-modifying therapies.
As this year marks the 200-year anniversary of the recognition of PD by James Parkinson4, we also celebrate the remarkable progress in understanding PD etiology and pathogenesis5. Here, we review recent advances in the study of the genetics, cell biology, and pathology of PD, highlighting emerging areas of overlap. Where these areas were previously studied in isolation, results from these disparate strands of research are beginning to converge, providing a more unified understanding of PD pathogenesis. We argue that this integrative approach to PD, in which seemingly disconnected results are re-examined as components of a cohesive whole, is also creating exciting opportunities for clinical applications.
Functional genomics and the promise of personalized medicine
Although only a small minority of patients with PD have thus far been found to have responsible pathogenic gene mutations, genetic discoveries have nevertheless been a driving force in the elucidation of PD mechanisms. Following on the early success from studies of rare families with Mendelian forms of PD, more than 20 PD genes and variants have now been implicated, including from large-scale genome-wide association studies (GWAS) and, more recently, whole exome sequencing (WES) studies in population-based cohorts6–8. A common challenge with either approach entails definitive confirmation of the responsible genes and elucidation of implicated disease mechanisms. In GWAS, implicated genomic regions often contain several equally plausible gene candidates. Whereas WES studies usually single out specific gene candidates, the implicated alleles may be too rare to definitively confirm a causal link to PD based on currently available sample sizes. Medium- to high-throughput screening assays in cellular or animal models can connect promising candidate genes to PD-relevant biologic mechanisms, prioritizing a subset for further study. For example, Jansen et al.9 evaluated 27 promising candidate genes with homozygous or compound heterozygous loss-of-function alleles based on WES in 1,148 unrelated young-onset PD cases. Since nearly all of the gene candidates were observed only once in the cohort, the investigators probed each gene for roles in mitochondrial dynamics or α-Syn-mediated toxicity using cellular and fruit fly experimental models. Ultimately, five genes were supported by both functional data and additional human genetic analyses consistent with replication. Beyond accelerating the discovery of novel PD genes, related approaches are also revealing the function of many other established genes/variants, grouping discrete susceptibility loci into common pathways and thereby consolidating our understanding of PD pathogenesis. For example, the recently identified PD gene CHCHD2 may mediate its activity through a mitochondrial pathway like other recessive PD genes (see below)10–12. Moreover, VPS35 and EIF4G1, both implicated in autosomal dominant forms of PD13–17, were recently found to genetically interact with one another and converge on α-Syn toxicity in yeast, worm, and mouse models of synucleinopathy18.
By contrast, with the sequence-based discovery of rare variant PD risk factors, the susceptibility alleles identified by GWAS7 usually do not alter protein-coding regions, making functional follow-up more challenging. For example, one of the earliest discovered PD risk polymorphisms at the human MAPT locus may primarily impact alternative mRNA splicing19,20. In another recent study, Soldner et al. used human pluripotent cell-derived neurons containing an intronic PD-related variant in the gene encoding α-Syn (SNCA)21. The authors found that this common polymorphism, which is present in about half of the population, coincided with a distal enhancer element resulting in an approximately 10% increase in SNCA transcript levels. It was therefore suggested that a mild increase of α-Syn over the course of decades renders individuals susceptible to PD. This is consistent with the findings from rare families with SNCA locus multiplication. In these cases, individuals with SNCA gene mulptiplication present with clinical features typical of PD, including a similar age at onset to sporadic PD22,23, whereas individuals with SNCA gene triplication present with a more early onset, aggressive form of PD24. Thus, SNCA gene dosage may be an integral feature of PD pathogenesis. In the case of the more common SNCA polymorphism, the modest increase in α-Syn protein levels may interact with other genetic risk variants or environmental exposures to cause PD. For example, Goldman et al. found that head injury was significantly associated with increased PD risk, but only in the context of a disease-associated SNCA promoter polymorphism25.
One of the great hopes for advances in PD genetics is to realize goals for personalized medicine, including improved risk prediction and even targeted therapies. It has long been speculated that much of PD’s clinical heterogeneity may be genetically encoded26,27. For example, besides their potent impact on risk of PD28, GBA mutations have been reported by several groups to cause an earlier age-at-onset, more rapid progression, and an increased risk of cognitive impairment and dementia in carriers29–34. Moreover, additional studies have looked at the effect of allelic heterogeneity on modifying PD clinical presentations35–37. In the future, it may be important to couple such studies examining the clinical impact of selected allelic variants with experimental investigations to define functional consequences in well-defined cellular or animal models. Moreover, once characterized, these models can serve as a platform for testing putative “personalized” treatments38–42. For example, Sanofi Genzyme is currently supporting a study (MOVES-PD) of GZ/SAR402671, a glucosylceramide synthase inhibitor, in PD patients carrying a GBA gene mutation (ClinicalTrials.gov, NCT02906020). Another study (AiM-PD) is examining the effects of oral Ambroxol, a glucocerebrosidase-modulating chaperone, in patients with PD (ClinicalTrials.gov, NCT02941822). Lastly, the use of biomarkers in stratifying clinical populations and understanding the biological underpinning of PD subtypes will be critically important when developing personalized medicine approaches. Specifically, profiling blood and CSF biomarkers may enhance disease subtyping based on clinical manifestations alone. This hypothesis is currently being tested in the Parkinson’s Progression Markers Initiative (PPMI) led by the Michael J. Fox Foundation for Parkinson’s Research43,44.
From genes to organelles and cellular homeostasis
The maintenance and function of cellular organelles, including mitochondria and lysosomes, are critical for functional neuronal integrity45–47. Interestingly, several previously identified PD genes such as PARK2, PARK6, and PARK7 (encoding Parkin, PTEN-induced putative kinase 1 [PINK1], and DJ-1, respectively) have been linked to mitochondrial function48–54. Since their initial discovery, a large body of work has elucidated a cellular pathway through which dysfunctional mitochondria can be recycled by way of autophagy or, more specifically, “mitophagy”55–59. Damaged mitochondria promote the phosphorylation of ubiquitin and Parkin by PINK1 and are subsequently degraded by the autophagosomal system59–62. Additionally, studies of these genes have provided insight into their mitochondrial functions in healthy and diseased contexts. For example, a recent study suggests that Parkin acts as an endogenous buffer for mitochondrial stress and its loss sensitizes dopaminergic neurons to mitochondrial mutations over time63,64. In addition, using a fly model, Vos and colleagues65 discovered a new pathway that could suppress the motor and biochemical abnormalities caused by PINK1 loss of function. Specifically, PINK1 genetically interacts with the enzyme responsible for the conversion of vitamin K1 into vitamin K265. Remarkably, supplementation of vitamin K2 could reverse PINK1 mutant phenotypes, suggesting a potential therapeutic approach. However, patient selection will be critical for potential clinical trials, as PARK6 mutations result in rare, autosomal recessive juvenile parkinsonism66, and it is uncertain whether a similar functional deficiency in vitamin K2 may apply in the general PD patient population. Identifying the subcellular impact of other PD genes may similarly lead to other targeted therapies.
Beyond discovering the primary targets of genetic abnormalities, it is essential to understand the subsequent cascade of cellular injury, such as how damaged mitochondria impact other cellular constituents, leading to neuronal dysfunction and death. In other words, discrete targets must be understood in the context of a cohesive, dynamic system. One example comes from recent studies that strongly implicate defects in the vesicular trafficking system67, which mediates cellular secretion and endocytosis as well as vesicle docking and fusion and is critically important for synaptic transmission, lysosomal degradation, and autophagy. Several PD genes, including VPS35, LRRK2, RAB7L1, GBA, and SNCA, have functions that converge on the vesicular trafficking system67–70. Leucine-rich repeat kinase 2 (LRRK2) and Rab-7-like protein 1 (RAB7L1), for example, act coordinately to regulate endolysosomal protein sorting via Rab GTPases, and several studies also support a key role for the cargo-shuttling retromer protein vacuolar protein sorting 35 (VPS35)71–73. In fact, the retromer is implicated as a critical downstream effector whose dysfunction may lead to neuronal toxicity and death74. Importantly, VPS35 may also play a role in mitophagy, perhaps via trafficking of mitochondria-derived vesicles to lysosomes75. The dense interconnections between PD genes and other regulators of vesicular trafficking were highlighted by recent work that combined an α-Syn protein–protein interaction network with suppressor-enhancer screening in yeast76,77. Based on these and other findings, chemical modulators of autophagy and/or the retromer, such as rapamycin (or similar “rapalogues”)78,79 and R5580,81, respectively, may be promising therapeutic avenues for targeting vesicular sorting defects in PD. However, since these pathways mediate essential functions in most tissues, successful dose titration to achieve selective action in the nervous system while minimizing potentially deleterious off-target effects is one anticipated challenge82. Nevertheless, these studies illustrate how the emerging, systems-based understanding of PD can highlight vulnerable “nodes” within complex cellular networks, creating promising therapeutic opportunities.
α-Syn toxicity and propagation: from cells to systems
The centrality of α-Syn in PD pathogenesis was established nearly two decades ago with the dual finding that (i) this protein is the principal constituent of the hallmark Lewy body pathology83 and (ii) SNCA gene mutations cause familial forms of PD84. Since then, α-Syn genomic locus multiplication22–24 or promoter polymorphisms that increase protein expression21 have also been confirmed as causal factors. Thus, intensive investigation has probed the relationship between α-Syn with PD pathogenesis. α-Syn was first described as a member of the synuclein family, which is associated with the synapse and the nucleus85. While studies on the physiological function of α-Syn suggest that it may play a role in synaptic transmission86 and aid in curving cellular membranes87, less is known regarding the mechanism(s) through which its gain-of-function causes neurodegeneration. Thus, one aspect of α-Syn research has focused on understanding how this protein causes toxicity within the cell. Studies in both animal models and human tissue have highlighted a role for α-Syn at the outer mitochondrial membrane88,89, the nucleus90–92, and the synapse93,94 as putative toxic mechanisms. Moreover, mechanisms involving proteostasis95,96 and lysosomal dysfunction45 that collectively lead to increased α-Syn levels are also tantalizing.
Given the direct relationship between α-Syn abundance and its role in PD pathogenesis and propagation, immunotherapy against α-Syn has emerged as one of the most promising therapeutic approaches for PD97,98. Results from ongoing and future immunotherapeutic trials by Prothena/Roche, Biogen, AFFiRiS, and other biotech companies will provide information on whether aggregated α-Syn is an important therapeutic target99. Nevertheless, caution is warranted given previous negative outcomes from immunotherapeutic trials of other neurodegenerative diseases, such as Alzheimer’s disease, characterized by protein aggregation (proteinopathies)100.
Another avenue for therapeutic intervention that is actively being investigated is based on the observation that hyperactivity of the non-receptor tyrosine kinase c-Abl contributes to α-Syn phosphorylation, accumulation, and neurodegeneration101. This has led to preliminary investigation of Nilotinib, a c-Abl inhibitor previously approved for the treatment of chronic myeloid leukemia, as a potential therapeutic agent for PD102. Further studies, however, are needed before this potent drug can be recommended as a symptomatic or disease-modifying therapy for PD103. A single-center trial at Georgetown University (ClinicalTrials.gov, NCT02954978) and a multicenter trial (NILO-PD) (ClinicalTrials.gov, NCT02281474), sponsored by the Michael J. Fox Foundation for Parkinson’s Research and the Parkinson Study Group, are currently under way.
Since the seminal observation by Braak and colleagues that most idiopathic PD cases fit a relatively predictable, caudal to rostral pathologic staging progression104, there has been great interest in understanding the mechanism by which α-Syn pathology may spread from the peripheral organs (e.g. the enteric or pericardial tissue) via the vagus nerve to the lower brainstem and eventually involve the neocortex. The intriguing possibility that α-Syn pathology can spread from cell to cell105–107 was suggested by observations of Lewy-like pathology in engrafted neurons from PD patients receiving fetal dopaminergic cell transplants108,109. First studies indicated that aggregates of α-Syn could enter cells that express transgenic α-Syn and seed the formation of new aggregates110,111. More recently, research findings have indicated that even synthetic, wild-type forms of α-Syn, if improperly folded and injected in the mouse brain, can induce the misfolding of otherwise normal endogenous α-Syn, thereby amplifying and propagating pathological forms112–114, properties consistent with those of prions (infectious proteinaceous agents)115. It appears that α-Syn can adopt a variety of different misfolded/aggregated oligomeric conformations that correspond to distinct profiles of toxicity in experimental assays116,117. These findings raise the intriguing, though yet unproven, hypothesis that certain α-Syn “strains” may contribute to clinical and pathologic heterogeneity among PD and other synucleinopathies. Interestingly, Mao et al.118 recently identified lymphocyte-activation gene 3 (LAG3) as a candidate receptor for α-Syn oligomeric “seeds”, and genetic manipulation of LAG3 in cells and mouse models altered pathologic progression. These findings raise the possibility of diagnostic and therapeutic advance based on the detection of α-Syn strains in patient populations as well as potential pharmacologic blockade of propagation118. It is also possible that neuroanatomic variation among key co-factors or receptors for α-Syn seeding or spread might contribute to the selective vulnerability of specific neuronal subpopulations in PD. Despite remarkable recent progress, the clinical relevance of α-Syn seeding and propagation (if any) remains to be fully understood105,119,120. For example, autopsies from some PD patients receiving fetal grafts were devoid of pathology two decades following transplantation121,122. Another recent study found that individuals receiving human growth hormone derived from human cadavers were at no greater risk of developing PD123. The extent of pathological spread of α-Syn does not always follow a defined trans-synaptic pattern nor does the brain areas it affects fully correlate with clinical measures105. Thus, a continued investigation into the mechanism through which α-Syn pathological assemblies form and how these are tied to toxicity is warranted.
Gastrointestinal dysmotility is a common early complaint in PD patients124,125, and the enteric nervous system has been implicated as an early target of α-Syn pathology104,126. Along with the growing interest in immunologic and inflammatory disease mechanisms, the gut microbiome has recently come under investigation in studies of both PD patients127–130 and animal models131. Sampson and colleagues131 found that α-Syn transgenic mice living in a germ-free environment were less vulnerable to neurodegeneration, but when the mice were inoculated with fecal bacteria taken from patients with PD their motor function deteriorated. While this study suggests the possibility that the gut microbiome may influence PD manifestations, it will be important to define the specific microbial contributors to the disease as well as recapitulate these findings in other disease models before moving forward into humans132. Beyond its potential role in disease modulation, the gastrointestinal tract might even be a trigger point for disease based on the findings of early enteric nervous system α-Syn pathology in some pathologic series104. Long before the current excitement concerning α-Syn propagation, Braak et al. speculated about a possible gastrointestinal pathogen (or other enteric exposure) as an initiating event, followed by pathologic spread via the vagus nerve to its medullary dorsal motor nucleus where PD changes first appear in the brainstem104,126,133. Following up on this provocative hypothesis, investigators recently found that subjects undergoing vagotomy (transection of the vagal nerve for the treatment of peptic ulcer disease) are at a modest but significantly reduced risk of PD134,135. While intriguing, the finding requires further confirmation, and animal model studies will be essential to definitively prove that the mechanism of protection is indeed based on the disruption of spread from the enteric to the central nervous system.
Conclusion
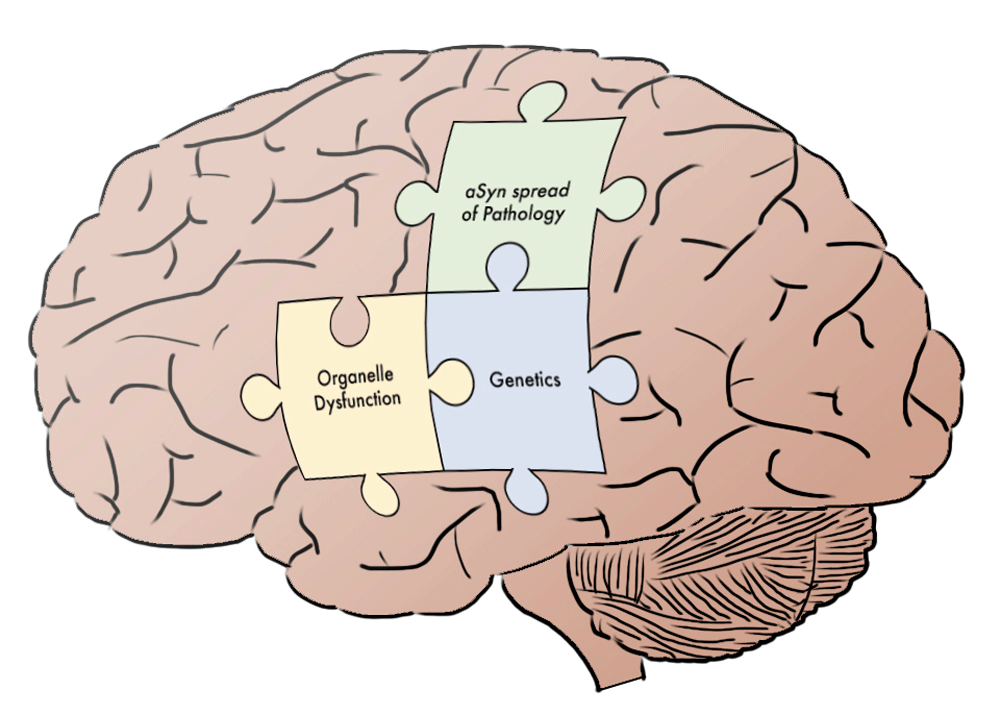
Recent advances have clearly enhanced our knowledge of the fundamental processes underlying PD pathogenesis. As connections are recognized among the disparate domains of PD inquiry, the broader patterns begin to emerge. As discussed above, we now recognize the cellular targets of many PD genes, such as how PARKIN and GBA regulate mitochondrial or lysosomal function, respectively. Additionally, the field has made progress toward understanding how dynamic interactions between such organelles impact overall cellular health, as in mitophagy. Lastly, studies of α-Syn illustrate the convergence of classical histopathologic analysis of PD with genetic investigations, and more recent investigations demonstrate how synucleinopathy not only impacts single cells or tissues but also may propagate throughout the nervous system. In sum, we are rapidly making progress toward a more cohesive model of PD pathogenesis, and this systems-level understanding is likely to accelerate therapeutic inroads. With the broad outlines of the “PD puzzle” now apparent (Figure 1), we predict that new insights can be more rapidly integrated within this framework. For example, forthcoming discoveries of new genetic risk loci can be understood within the context of known functional pathways within both neurons and other cell types, such as astrocytes or microglia. An integrated understanding of PD will also enable more effective multi- and inter-disciplinary collaboration among scientists and clinicians, driving next-generation therapeutic trials targeting disease mechanisms and fulfilling the promise of personalized medicine. Advances in understanding the cellular mechanism underlying PD-related neurodegeneration will undoubtedly lead to better symptomatic and novel pathogenesis-targeted, disease-modifying therapies136.
Figure 1. Putting the Parkinson’s disease (PD) puzzle together.
Three pieces of the “PD puzzle” covered in this short review are highlighted within the context of the brain. Additional elements remain to be fully elucidated. The integrated, systems-level understanding of PD that emerges from understanding how these components fit together will likely accelerate therapeutic advances. α-Syn, α-synuclein.
Maxime W.C. Rousseaux and Joshua M. Shulman declare that they have no disclosures. Joseph Jankovic has received research or training grants and/or has served as a consultant for the Michael J. Fox Foundation for Parkinson’s Research, Prothena Biosciences Inc, and Teva Pharmaceutical Industries Ltd.
Grant information
Maxime W.C. Rousseaux is supported in part by Grant No. PF-JFA-1762 from the Parkinson’s Disease Foundation. Joshua M. Shulman is supported by the Huffington Foundation, the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital, and a Career Award for Medical Scientists from the Burroughs Wellcome Fund. Joseph Jankovic has received research and/or educational support from the Michael J. Fox Foundation for Parkinson’s Research, Parkinson’s Foundation, the Parkinson Study Group, Prothena Biosciences Inc, and Teva Pharmaceutical Industries Ltd.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
2.
Postuma RB, Berg D, Stern M, et al.:
MDS clinical diagnostic criteria for Parkinson’s disease.
Mov Disord.
2015; 30(12): 1591–601. PubMed Abstract
| Publisher Full Text
3.
Lotia M, Jankovic J:
New and emerging medical therapies in Parkinson’s disease.
Expert Opin Pharmacother.
2016; 17(7): 895–909. PubMed Abstract
| Publisher Full Text
4.
Parkinson J:
An essay on the shaking palsy. 1817.
J Neuropsychiatry Clin Neurosci.
2002; 14(2): 223–36; discussion 222. PubMed Abstract
| Publisher Full Text
5.
Jankovic J:
Movement disorders in 2016: progress in Parkinson disease and other movement disorders.
Nat Rev Neurol.
2017; 13(2): 76–8. PubMed Abstract
| Publisher Full Text
7.
Nalls MA, Pankratz N, Lill CM, et al.:
Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease.
Nat Genet.
2014; 46(9): 989–93. PubMed Abstract
| Publisher Full Text
| Free Full Text
9.
Jansen IE, Ye H, Heetveld S, et al.:
Discovery and functional prioritization of Parkinson’s disease candidate genes from large-scale whole exome sequencing.
Genome Biol.
2017; 18(1): 22. PubMed Abstract
| Publisher Full Text
| Free Full Text
16.
Deng H, Wu Y, Jankovic J:
The EIF4G1 gene and Parkinson’s disease.
Acta Neurol Scand.
2015; 132(2): 73–8. PubMed Abstract
| Publisher Full Text
17.
Chartier-Harlin MC, Dachsel JC, Vilariño-Güell C, et al.:
Translation initiator EIF4G1 mutations in familial Parkinson disease.
Am J Hum Genet.
2011; 89(3): 398–406. PubMed Abstract
| Publisher Full Text
| Free Full Text
19.
Trabzuni D, Wray S, Vandrovcova J, et al.:
MAPT expression and splicing is differentially regulated by brain region: relation to genotype and implication for tauopathies.
Hum Mol Genet.
2012; 21(18): 4094–103. PubMed Abstract
| Publisher Full Text
| Free Full Text
20.
Valenca GT, Srivastava GP, Oliveira-Filho J, et al.:
The role of MAPT haplotype H2 and isoform 1N/4R in parkinsonism of older adults.
PLoS One.
2016; 11(7): e0157452. PubMed Abstract
| Publisher Full Text
| Free Full Text
22.
Chartier-Harlin MC, Kachergus J, Roumier C, et al.:
Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease.
Lancet.
2004; 364(9440): 1167–9. PubMed Abstract
| Publisher Full Text
23.
Ibáñez P, Bonnet AM, Débarges B, et al.:
Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease.
Lancet.
2004; 364(9440): 1169–71. PubMed Abstract
| Publisher Full Text
28.
Sidransky E, Nalls MA, Aasly JO, et al.:
Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease.
N Engl J Med.
2009; 361(17): 1651–61. PubMed Abstract
| Publisher Full Text
| Free Full Text
31.
Brockmann K, Srulijes K, Pflederer S, et al.:
GBA-associated Parkinson’s disease: reduced survival and more rapid progression in a prospective longitudinal study.
Mov Disord.
2015; 30(3): 407–11. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
32.
Clark LN, Ross BM, Wang Y, et al.:
Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease.
Neurology.
2007; 69(12): 1270–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
34.
Winder-Rhodes SE, Evans JR, Ban M, et al.:
Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort.
Brain.
2013; 136(Pt 2): 392–9. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
36.
Pihlstrøm L, Morset KR, Grimstad E, et al.:
A cumulative genetic risk score predicts progression in Parkinson’s disease.
Mov Disord.
2016; 31(4): 487–90. PubMed Abstract
| Publisher Full Text
37.
Mata IF, Leverenz JB, Weintraub D, et al.:
GBA variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease.
Mov Disord.
2016; 31(1): 95–102. PubMed Abstract
| Publisher Full Text
| Free Full Text
38.
Migdalska-Richards A, Daly L, Bezard E, et al.:
Ambroxol effects in glucocerebrosidase and α-synuclein transgenic mice.
Ann Neurol.
2016; 80(5): 766–75. PubMed Abstract
| Publisher Full Text
| Free Full Text
39.
Ishay Y, Zimran A, Szer J, et al.:
Combined beta-glucosylceramide and ambroxol hydrochloride in patients with Gaucher related Parkinson disease: from clinical observations to drug development.
Blood Cells Mol Dis.
2016; pii: S1079-9796(16)30229-7. PubMed Abstract
| Publisher Full Text
40.
Migdalska-Richards A, Ko WKD, Li Q, et al.:
Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate.
Synapse.
2017; 71(7). PubMed Abstract
| Publisher Full Text
42.
Zhang YS, Aleman J, Shin SR, et al.:
Multisensor-integrated organs-on-chips platform for automated and continual in situ monitoring of organoid behaviors.
Proc Natl Acad Sci U S A.
2017; 114(12): E2293–E2302. PubMed Abstract
| Publisher Full Text
| Free Full Text
43.
Espay AJ, Schwarzschild MA, Tanner CM, et al.:
Biomarker-driven phenotyping in Parkinson’s disease: a translational missing link in disease-modifying clinical trials.
Mov Disord.
2017; 32(3): 319–24. PubMed Abstract
| Publisher Full Text
| Free Full Text
44.
Parkinson Progression Marker Initiative:
The Parkinson Progression Marker Initiative (PPMI).
Prog Neurobiol.
2011; 95(4): 629–35. PubMed Abstract
| Publisher Full Text
45.
Manzoni C, Lewis PA:
Dysfunction of the autophagy/lysosomal degradation pathway is a shared feature of the genetic synucleinopathies.
FASEB J.
2013; 27(9): 3424–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
47.
Van Laar VS, Berman SB:
The interplay of neuronal mitochondrial dynamics and bioenergetics: implications for Parkinson’s disease.
Neurobiol Dis.
2013; 51: 43–55. PubMed Abstract
| Publisher Full Text
| Free Full Text
49.
Irrcher I, Aleyasin H, Seifert EL, et al.:
Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics.
Hum Mol Genet.
2010; 19(19): 3734–46. PubMed Abstract
| Publisher Full Text
52.
Yang Y, Gehrke S, Imai Y, et al.:
Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila pink1 is rescued by parkin.
Proc Natl Acad Sci U S A.
2006; 103(28): 10793–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
53.
Canet-Avilés RM, Wilson MA, Miller DW, et al.:
The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization.
Proc Natl Acad Sci U S A.
2004; 101(24): 9103–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
54.
Andres-Mateos E, Perier C, Zhang L, et al.:
DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase.
Proc Natl Acad Sci U S A.
2007; 104(37): 14807–12. PubMed Abstract
| Publisher Full Text
| Free Full Text
70.
Lashuel HA, Hirling H:
Rescuing defective vesicular trafficking protects against alpha-synuclein toxicity in cellular and animal models of Parkinson’s disease.
ACS Chem Biol.
2006; 1(7): 420–4. PubMed Abstract
| Publisher Full Text
73.
Beilina A, Rudenko IN, Kaganovich A, et al.:
Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease.
Proc Natl Acad Sci U S A.
2014; 111(7): 2626–31. PubMed Abstract
| Publisher Full Text
| Free Full Text
77.
Chung CY, Khurana V, Yi S, et al.:
In situ peroxidase labeling and mass-spectrometry connects alpha-synuclein directly to endocytic trafficking and mRNA metabolism in neurons.
Cell Syst.
2017; 4(2): 242–250.e4. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
78.
Malagelada C, Jin ZH, Jackson-Lewis V, et al.:
Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease.
J Neurosci.
2010; 30(3): 1166–75. PubMed Abstract
| Publisher Full Text
| Free Full Text
79.
Moors TE, Hoozemans JJ, Ingrassia A, et al.:
Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease.
Mol Neurodegener.
2017; 12(1): 11. PubMed Abstract
| Publisher Full Text
| Free Full Text
80.
Follett J, Bugarcic A, Yang Z, et al.:
Parkinson disease-linked Vps35 R524W mutation impairs the endosomal association of retromer and induces α-synuclein aggregation.
J Biol Chem.
2016; 291(35): 18283–98. PubMed Abstract
| Publisher Full Text
| Free Full Text
83.
Spillantini MG, Schmidt ML, Lee VM, et al.:
Alpha-synuclein in Lewy bodies.
Nature.
1997; 388(6645): 839–40. PubMed Abstract
| Publisher Full Text
84.
Polymeropoulos MH, Lavedan C, Leroy E, et al.:
Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease.
Science.
1997; 276(5321): 2045–7. PubMed Abstract
| Publisher Full Text
85.
Maroteaux L, Campanelli JT, Scheller RH:
Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal.
J Neurosci.
1988; 8(8): 2804–15. PubMed Abstract
86.
Wales P, Pinho R, Lázaro DF, et al.:
Limelight on alpha-synuclein: pathological and mechanistic implications in neurodegeneration.
J Parkinsons Dis.
2013; 3(4): 415–59. PubMed Abstract
| Publisher Full Text
91.
Rousseaux MW, de Haro M, Lasagna-Reeves CA, et al.:
TRIM28 regulates the nuclear accumulation and toxicity of both alpha-synuclein and tau.
eLife.
2016; 5: pii: e19809. PubMed Abstract
| Publisher Full Text
| Free Full Text
94.
Garcia-Reitböck P, Anichtchik O, Bellucci A, et al.:
SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson’s disease.
Brain.
2010; 133(Pt 7): 2032–44. PubMed Abstract
| Publisher Full Text
| Free Full Text
95.
Schildknecht S, Gerding HR, Karreman C, et al.:
Oxidative and nitrative alpha-synuclein modifications and proteostatic stress: implications for disease mechanisms and interventions in synucleinopathies.
J Neurochem.
2013; 125(4): 491–511. PubMed Abstract
| Publisher Full Text
96.
Cook C, Stetler C, Petrucelli L:
Disruption of protein quality control in Parkinson’s disease.
Cold Spring Harb Perspect Med.
2012; 2(5): a009423. PubMed Abstract
| Publisher Full Text
| Free Full Text
97.
Valera E, Spencer B, Masliah E:
Immunotherapeutic approaches targeting amyloid-β, α-synuclein, and tau for the treatment of neurodegenerative disorders.
Neurotherapeutics.
2016; 13(1): 179–89. PubMed Abstract
| Publisher Full Text
| Free Full Text
99.
Jankovic J, Godman I, Safirstein B, et al.:
Results from a phase 1b multiple ascending-dose study of PRX002, an anti-alpha-synuclein monoclonal antibody, in patients with Parkinson’s disease.
Neurodegener Dis.
2017; 17(suppl 1)(567). [Abstract].
100.
Wang Y:
Alzheimer disease: lessons from immunotherapy for Alzheimer disease.
Nat Rev Neurol.
2014; 10(4): 188–9. PubMed Abstract
| Publisher Full Text
103.
Robledo I, Jankovic J:
Media hype: patient and scientific perspectives on misleading medical news.
Mov Disord.
2017. PubMed Abstract
| Publisher Full Text
104.
Braak H, Del Tredici K, Rüb U, et al.:
Staging of brain pathology related to sporadic Parkinson’s disease.
Neurobiol Aging.
2003; 24(2): 197–211. PubMed Abstract
| Publisher Full Text
106.
Hasegawa M, Nonaka T, Masuda-Suzukake M:
α-Synuclein: experimental pathology.
Cold Spring Harb Perspect Med.
2016; 6(9): pii: a024273. PubMed Abstract
| Publisher Full Text
107.
Luna E, Luk KC:
Bent out of shape: α-synuclein misfolding and the convergence of pathogenic pathways in Parkinson’s disease.
FEBS Lett.
2015; 589(24 Pt A): 3749–59. PubMed Abstract
| Publisher Full Text
| Free Full Text
108.
Kordower JH, Chu Y, Hauser RA, et al.:
Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease.
Nat Med.
2008; 14(5): 504–6. PubMed Abstract
| Publisher Full Text
111.
Luk KC, Song C, O'Brien P, et al.:
Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells.
Proc Natl Acad Sci U S A.
2009; 106(47): 20051–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
119.
McCann H, Cartwright H, Halliday GM:
Neuropathology of α-synuclein propagation and braak hypothesis.
Mov Disord.
2016; 31(2): 152–60. PubMed Abstract
| Publisher Full Text
120.
Hansen C, Li JY:
Beyond α-synuclein transfer: pathology propagation in Parkinson’s disease.
Trends Mol Med.
2012; 18(5): 248–55. PubMed Abstract
| Publisher Full Text
121.
Hallett PJ, Cooper O, Sadi D, et al.:
Long-term health of dopaminergic neuron transplants in Parkinson’s disease patients.
Cell Rep.
2014; 7(6): 1755–61. PubMed Abstract
| Publisher Full Text
| Free Full Text
122.
Mendez I, Viñuela A, Astradsson A, et al.:
Dopamine neurons implanted into people with Parkinson’s disease survive without pathology for 14 years.
Nat Med.
2008; 14(5): 507–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
123.
Irwin DJ, Abrams JY, Schonberger LB, et al.:
Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone.
JAMA Neurol.
2013; 70(4): 462–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
124.
Su A, Gandhy R, Barlow C, et al.:
A practical review of gastrointestinal manifestations in Parkinson’s disease.
Parkinsonism Relat Disord.
2017; 39: 17–26. PubMed Abstract
| Publisher Full Text
125.
Postuma RB, Berg D:
Advances in markers of prodromal Parkinson disease.
Nat Rev Neurol.
2016; 12(11): 622–34. PubMed Abstract
| Publisher Full Text
126.
Braak H, Rüb U, Gai WP, et al.:
Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen.
J Neural Transm (Vienna).
2003; 110(5): 517–36. PubMed Abstract
| Publisher Full Text
130.
Wood H:
Parkinson disease. Gut reactions--can changes in the intestinal microbiome provide new insights into Parkinson disease?
Nat Rev Neurol.
2015; 11(2): 66. PubMed Abstract
| Publisher Full Text
132.
Parashar A, Udayabanu M:
Gut microbiota: implications in Parkinson’s disease.
Parkinsonism Relat Disord.
2017; 38: 1–7. PubMed Abstract
| Publisher Full Text
133.
Braak H, Del Tredici K:
Neuropathological staging of brain pathology in sporadic Parkinson’s disease: separating the wheat from the chaff.
J Parkinsons Dis.
2017; 7(s1): S73–S87. PubMed Abstract
| Publisher Full Text
| Free Full Text
1
Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, 1250 Moursund St, Houston, TX, 77030, USA 2
Department of Molecular and Human Genetics, Baylor College of Medicine, One Baylor Plaza, Houston, TX, 77030, USA 3
Parkinson's Disease Center and Movement Disorders Clinic, Department of Neurology, Baylor College of Medicine, 7200 Cambridge, Houston, TX, 77030-4202, USA 4
Department of Neuroscience, Baylor College of Medicine, One Baylor Plaza, Houston, TX, 77030, USA
Maxime W.C. Rousseaux
Roles:
Writing – Original Draft Preparation,
Writing – Review & Editing
Joshua M. Shulman
Roles:
Writing – Original Draft Preparation,
Writing – Review & Editing
Joseph Jankovic
Roles:
Writing – Original Draft Preparation,
Writing – Review & Editing
Maxime W.C. Rousseaux and Joshua M. Shulman declare that they have no disclosures. Joseph Jankovic has received research or training grants and/or has served as a consultant for the Michael J. Fox Foundation for Parkinson’s Research, Prothena Biosciences Inc, and Teva Pharmaceutical Industries Ltd.
Maxime W.C. Rousseaux is supported in part by Grant No. PF-JFA-1762 from the Parkinson’s Disease Foundation. Joshua M. Shulman is supported by the Huffington Foundation, the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital, and a Career Award for Medical Scientists from the Burroughs Wellcome Fund. Joseph Jankovic has received research and/or educational support from the Michael J. Fox Foundation for Parkinson’s Research, Parkinson’s Foundation, the Parkinson Study Group, Prothena Biosciences Inc, and Teva Pharmaceutical Industries Ltd.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Rousseaux MWC, Shulman JM and Jankovic J. Progress toward an integrated understanding of Parkinson’s disease [version 1; peer review: 2 approved]. F1000Research 2017, 6(F1000 Faculty Rev):1121 (https://doi.org/10.12688/f1000research.11820.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)