Keywords
HITS-CLIP, CLIP-SEQ, peak caller
HITS-CLIP, CLIP-SEQ, peak caller
A peak caller that could accurately define a single peak amongst several samples was required to analyze High-Throughput Sequencing of RNA isolated by Crosslinking Immunoprecipitation (HITS-CLIP) (Darnell, 2010) data from fetal liver red blood cell precursors of miR-144/451-/- and wild-type mice (Paralkar et al., 2014; Paralkar VR, Palmer LE, Xu P, Lechauve C, Zhao G, Yao Y, Luan J, Wu G, Vourekas A, Mourelatos Z, Scheutz JD and Weiss MJ; unpublished study). Piranha (Uren et al., 2012) is one such software that is commonly used to identify peaks generated by HITS-CLIP. However, Piranha bins the starts of reads and does not fully define a peak. Consequently, a large bin size may result in multiple peaks being combined into one peak. We found that the identification and resolution of peaks using Piranha was highly dependent on the background threshold (-a) and binSize (-b) parameters, and it was unclear how these parameters should be set in order to obtain the most biologically relevant information. We also found that running Piranha on multiple samples separately results in peak boundaries that may be quite different from sample to sample. Generating initial peak calls from a combined sample dataset creates a single standard set of peak boundaries for all samples, which simplifies downstream analysis. We therefore developed a peak calling algorithm, named YODEL, with the following properties: 1) Incorporate strand specificity (Piranha does this, but many other CHIP-SEQ peak callers do not); 2) Generate per-sample read counts for each peak; 3) Have parameters that have easily understandable implications when changed.
The main input for the peak caller is a BED file generated by clusterBed from the BEDtools suite (Quinlan & Hall, 2010) with the -s option used (see Supplementary material: Input file formats). If multiple samples are to be analyzed simultaneously, the name field must contain the sample name or ID before the first colon, followed by the read ID or other descriptive text. In addition, a sample list must be designated (with the YODEL parameter -sampleList) (see Supplementary material: Input file formats). The sample list will identify which samples are to be included for peak calling. After peak calling, read counts for each peak in all samples will be calculated. If no sample list is provided, all reads will be treated as one sample. As an example of how to process HITS-CLIP FASTQ files to generate the input clustered BED file, see Supplementary material: Analysis of HITS-CLIP data from Chi et al., 2009.
YODEL was written in Python and tested with Python version 2.7. YODEL processes each read cluster as it is encountered within the input clustered BED file. For each cluster, the base coverage at each position for all samples under examination for peak calling is determined. Position specific counts for all individual samples are calculated as well. Once the counts are tallied, the program iteratively identifies peaks until no additional peaks are found. The program identifies the position with the highest read count. If the read count is less than the minimum peak height (mph) than no further peak calling for the cluster is performed. From the position with the highest read count, bases upstream are analyzed one at a time. The lowest read count (lowestPoint) up to the current base is tracked. Where dipTolerance (dt) and peakDipBuffer (pdb) are input parameters, if at any position the count is 0 or the count >= (lowestPoint+ peakDipBuffer)* dipTolerance, then the peak start has been determined and is recorded as the base position where the count was 0 or the base position of lowestPoint. This is repeated for bases downstream of the highest point to find the peak end. The peak summit is defined as the median of all the positions with the highest count. Two sets of peak boundaries (25% and 50%) are defined as the positions where the coverage is 25% and 50% of the highest point, and the maximum peak heights per sample are determined. The numbers of reads per peak are calculated (at both the 25% and 50% range) by determining the number of reads that overlap the peak by at least the input parameter binSize (bs). Typically, we have used the 25% peak boundary for downstream calculations. The peak counts are determined on a per sample basis, and for all the samples used in peak calling combined. Once a peak has been determined, the base coverage for positions within the full peak is set to 0 so that no further peaks are called overlapping it. The peak finding process (starting with finding the position with highest read count) is repeated until no more peaks are identified. After peaks are identified by YODEL, several filtering steps can be applied to remove low quality peaks (see Supplementary material: Peak filtering, Supplementary Figure 2–Supplementary Figure 6).
YODEL has been tested on both Windows and Linux running Python 2.7 with standard libraries. Some of the tools (e.g. Bedtools) used to generate input files are Linux or OS X specific. There is no minimum memory requirement for YODEL, but the size of any BED files sorted with the Linux sort command may be dependent on system memory. See ‘Supplementary material: Analysis of HITS-CLIP data from Chi et al., 2009’ for instructions on how to preprocess data and run YODEL.
The output of YODEL is described in the Table 1. Figure 1 shows a comparison of YODEL and Piranha output from HITS-CLIP analysis of wild type and miR144/451-/- fetal liver erythroblasts. Results from two different YODEL parameter settings are shown in blue in the lower half of the figure. Cab39 (Figure 1A) (Godlewski et al., 2010) and Ywhaz (Figure 1B) (Yu et al., 2010) are two known miR-451a target mRNAs. For Cab39, the largest peak contains a miR-451a seed match that is not present in the knockout sample. Because Piranha bins the start of reads, the peak defined by Piranha may not actually include the seed match location, as is observed with the Cab39 seed match. The failure of a peak to cover a seed match may prevent a microRNA from being assigned to a peak and subsequently interfere with downstream analysis. Also, the peak calling of Piranha is greatly influenced by bin size. A bin size of 32 (default parameter) is not able to resolve many individual peaks. In Ywhaz mRNA (panel B) YODEL detects three peaks around the miR-451a seed match. One Piranha setting (a=0.98, b=16) identified the three individual peaks, but compromised detection of smaller peaks upstream of the predicted miR-451a binding site. Therefore for our miR144/451-/- data set, YODEL was superior to Piranha in defining HITS-CLIP peaks.
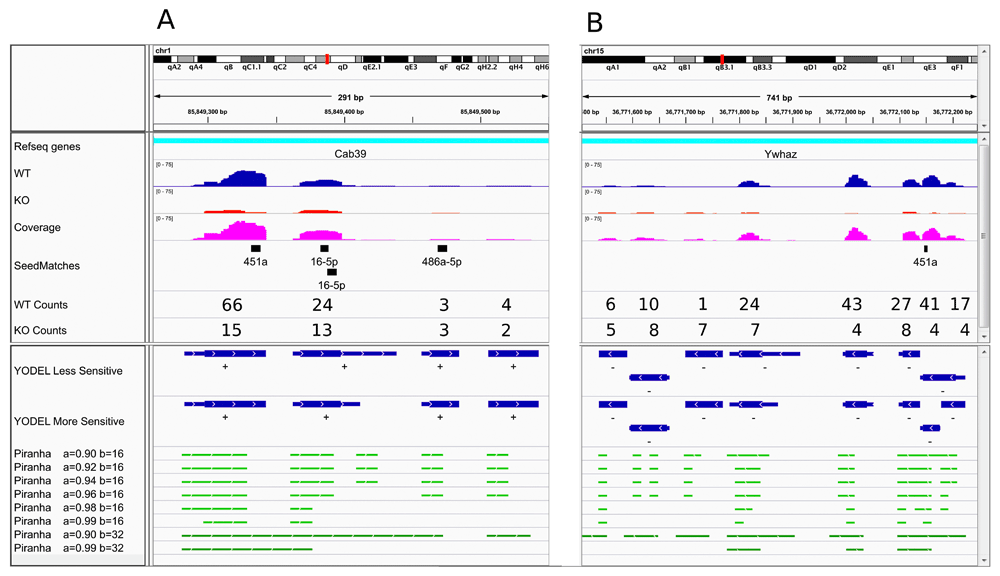
IGV browser (Thorvaldsdóttir et al., 2013) images showing YODEL output for HITS-CLIP data analyzing the 3’ untranslated regions of Cab39 (A) and Ywhaz (B) mRNAs in wild-type and miR-451a/miR-144-/- mouse fetal livers at embryonic day 14.5. The coverage tracks (WT in blue, KO in red, and combined coverage in magenta) show combined sequencing reads from three animals of each genotype mapped to the mouse mm10 genome. The seedMatches track shows microRNA seed matches for miR-451a, miR-144-3p and the three most abundant erythroid microRNAs besides miR-451a (miR-16-5p, miR-486a-5p and miR-122-5p) (Paralkar VR, Palmer LE, Xu P, Lechauve C, Zhao G, Yao Y, Luan J, Wu G, Vourekas A, Mourelatos Z, Scheutz JD and Weiss MJ; unpublished results). Average peak counts per sample are shown for wild-type and miR-451a/miR-144-/- erythroblasts using the YODEL more sensitive parameters (see below). The lower panels show YODEL (blue) and Piranha (green) peak boundaries with the indicated parameter settings. For YODEL, full boundaries are shown by thin lines and 25% boundaries by thick lines. YODEL less sensitive parameter settings: dipTolerance=2, peakDipBuffer=2; more sensitive settings: dipTolerance=1.5, peakDipBuffer=1; Both parameter settings: binSize=16, minPeakHeight=5. Note that the ability of Piranha to resolve different peaks representing distinct microRNA binding sites is highly dependent on parameter settings. Piranha parameters: a=background, threshold b=binSize. The default for Piranha is a=0.99 and b=32.
We have also tested the YODEL software on a publically available HITS-CLIP data set. HITS-CLIP reads from mouse neocortex Argonaute immunoprecipitations (Chi et al., 2009) was retrieved from http://ago.rockefeller.edu/rawdata.php. The reads were pre-processed and run through YODEL, as described in the Supplementary material: Analysis of HITS-CLIP data from Chi et al., 2009. We examined the first four genes identified by Table 1 in Chi et al., as potential target of microRNAs. Figure 2 shows a comparison of peak calling in the 3’ UTR of the Plod3 gene. Again, it is seen that binning starts of reads will cause potential microRNA seeds to be missed. Figure 3 shows the 3’ UTR of the Cd164 gene. See Supplementary Figure 1 for browser images of Ctdsp1 and Itgb1.

IGV browser image of a portion of the Plod3 3’ UTR. Mouse neocortex HITS-CLIP read coverage is shown along with peak calls from YODEL (lower panel blue bars) and Piranha (lower panel green bars). Relevant microRNA seeds (for seeds mapped see Supplementary material: Seeds mapped for brain neocortex data) are shown in black. For YODEL, full boundaries are shown by thin lines and 25% boundaries by thick lines. YODEL parameters: dipTolerance=1.5, peakDipBuffer=1, binSize=16, minPeakHeight=5. Piranha parameters: a=background, threshold b=binSize. The default for Piranha is a=0.99 and b=32.
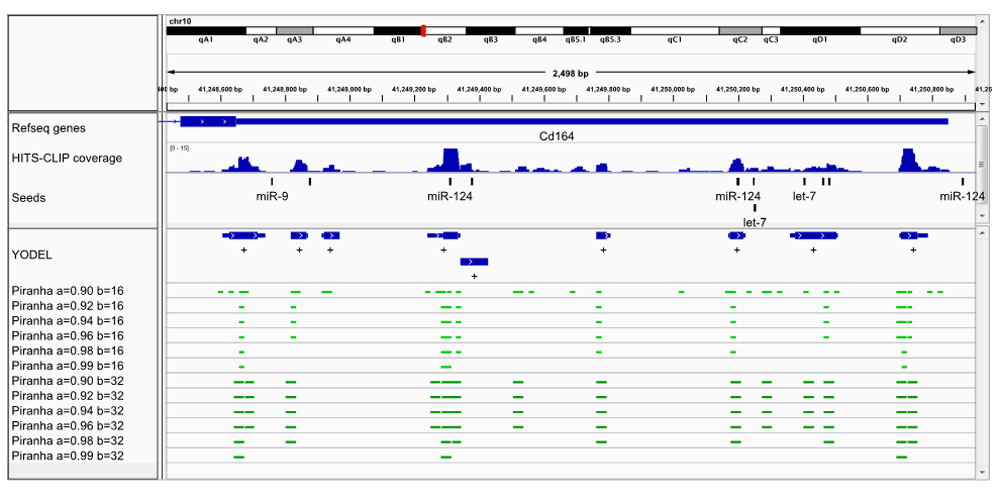
IGV browser image of a portion of the Cd164 3’ UTR. Mouse neocortex HITS-CLIP read coverage is shown along with peak calls from YODEL (lower panel blue bars) and Piranha (lower panel green bars). Relevant microRNA seeds (For seeds mapped see Supplementary material: Seeds mapped for brain neocortex data) are shown in black. For YODEL, full boundaries are shown by thin lines and 25% boundaries by thick lines. YODEL parameters: dipTolerance=1.5, peakDipBuffer=1, binSize=16, minPeakHeight=5. Piranha parameters: a=background, threshold b=binSize. The default for Piranha is a=0.99 and b=32.
We have designed a new peak-caller, termed YODEL, for analysis of RNA-seq data generated by HITS-CLIP-type experiments. Advantages of YODEL compared to Piranha, a program commonly used for the same purpose, include standardization of peak calls for comparative analysis of multiple samples, improved resolution of peak boundaries, and more consistent overlap between peak calls and microRNA seed matches.
YODEL is a Python script and is available at: https://github.com/LancePalmerStJude/YODEL/
Archived source code as at time of publication: https://doi.org/10.5281/zenodo.820635 (Palmer, 2017).
License: GPLv3
Input BED files for peak calling with the miR-144/451 KO HITS-CLIP can be found in the Cab39_Ywhaz.allSamples.fullCollapsed.clusters.bed file within the archived source code listed above. This file contains the clustered BED file used for YODEL input (only around Cab39 and Ywhaz).
Sample HITS-CLIP data from the 130kD band from mouse neocortex samples was downloaded from http://ago.rockefeller.edu/rawdata.php (130kD Brain A-E samples).
The pre-processing BASH pipeline used to generate a clustered BED files, as well as some accessory scripts, can be found in the Ch2009 directory within the archived source code link listed above.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)