Keywords
Phenotype simulation, GWAS
Phenotype simulation, GWAS
Genome-wide association studies (GWAS) are routine in population research. New methods are being developed for better accessing complex associations between genotypes and phenotypes, uncovering genotype structures or testing evolutionary hypotheses. Testing the novel methods requires experimental data, which may not be easily obtainable. One solution is to use artificial data simulated with specific assumptions.
The best existing phenotype simulators, such as: GENOME1, Plink2, phenosim3, CoaSim4, Fregene5, ForSim6, QuantiNemo7, GCTA8, HapGen9, SeqSimla10, and SimRare11 offer qualitative and dichotomous simulated phenotype. But the known phenotype simulation software tools have some limitations, which may prevent customers from using them: (i) the majority, if not all, of the phenotype simulation software tools do not offer simulation of survival traits/time-to-event outcome, making it impossible to test respective hypotheses of associations; (ii) some of the tools are not easy to use, due to wide range of parameters, which the user has to provide and control (rather than calculate them automatically), making them unnecessarily difficult to use and preventing the user from future use of the tool; (iii) phenotype simulation is often offered as an auxiliary part of the genetic simulation routine, and therefore the user first has to perform a time-consuming unavoidable genetic simulation in order to obtain the phenotype; (iv) in situations when the genetic data is already simulated from other tools, only phenosim and GCTA offer adding simulated phenotype to such data. Consequently, it is necessary to have a new, simple and flexible phenotype simulation tool with plain algorithmic assumptions.
Consequently, we present cophesim, a comprehensive phenotype simulation tool that was developed to add a phenotype to corresponding genotypes simulated by other simulation tool (Table S1). cophesim offers simulation of continuous, dichotomous and survival traits, with different (user-provided) effect sizes of causal variants, with the ability to simulate epistatic interactions. It also can simulate phenotype within gene-environment interaction assumptions using up to 10 covariates.
The workflow (see Figure 1) includes the following stages: (i) Input data pre-processing; (ii) phenotype simulation; (iii) generation of final output files.
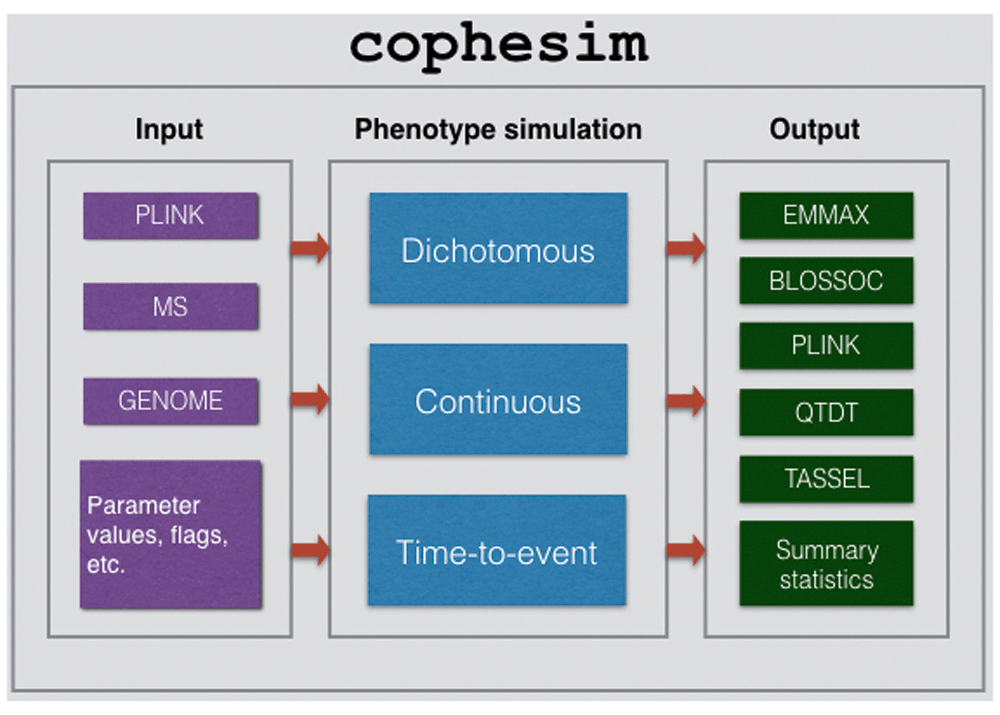
Workflow of cophesim has three stages: (1) Input stage, where the input data (can be provided in one of the three formats: Plink, MS and GENOME, see the user manual - Supplementary File 1) along with the other input parameters (such as causal variants with size effects, output format, etc.) is prepared for phenotype simulation; (2) Phenotype simulation stage, where different types of phenotypic traits are simulated: dichotomous, continuous and time-to-event (‘survival’); (3) Output stage – the final stage, where simulated phenotype data are packed to various formats in order to be directly usable by six GWAS tools: EMMAX, BLOSSOC, Plink, QTDT, TASSEL and GenABEL. Summary statistics are generated at the output stage as well.
Currently cophesim accepts the genotype output data from Plink, MS12 and GENOME software applications. Phenotypes (dichotomous, continuous and survival) are then added according to the following simulation scenarios.
Dichotomous phenotype for ith individual (i = 1...N, where N is the total number of individuals in a dataset) is simulated according to the logistic model (if the user provided effect sizes for causal variants):
where pi is the probability of a particular outcome. In cophesim, it is a probability of a “case” (cases are marked by “1”, and “0” are controls in simulated dichotomous phenotype) for ith individual. If pi is greater than the some threshold p0 (we use p0 ~ U(0, 1)), then the phenotype for ith individual is set to “1” and to “0” otherwise. The variable z is determined with the following equation: Ej – effect size for jth variant, user-defined; gij – value of jth genetic marker for ith individual; αj - effect size for jth covariate and Xij is a value of jth covariate for a ith individual (the term is added to represent gene-environment iterations); ϵi – a standard normal residual, ϵi N (0, 1), computed for ith individual, M is a total number of genetic variants and K is a total number of covariates used.If the user did not provide the effect sizes for causal variants, the following strategy is then used:
Here wij is a weight and computed as follows: (a standardization procedure, and the matrix W containing element wij is called a standardized genotype matrix8; MAFj – a minor allele frequency for jth genetic variant, and the other values are the same as described above. This strategy allows using defined genetic architecture in a simulated population.
Qualitative (continuous) phenotype for ith individual is simulated according to the linear regression scenario according to the equations (2) or (3) (in case if effect sizes were not supplied).
We model a survival phenotype from the proportional hazards model using the inverse probability method13: if U is uniform in (0, 1) and if S(·|z) is the conditional survival function derived from the proportional hazards model: S(t|z) = e–H0(t)ez, then the random variable
has survival function S(·|z). In this equation, H0(t) is a cumulative baseline hazard. By default, we use the Weibull cumulative baseline hazard: H0(t) = λρtρ–1; z is the same parameter that defined above, for each individual, and depends on whether the user provided effect sizes for causal variants or not. We also implemented exponential and Gompertz hazards.The simplest way to simulate collinearity between two SNPs, g1 and g2, with effect sizes E1 and E2 is to replace some portion of g2 with g1 values according to provided coefficient, which reflects a correlation between two SNPs. We also consider applying other techniques, such as copulas, in order to simulate LD.
These are modeled with the following equation for ith individual:
where the term E12 g1i g2i is the interaction term in which E12 is the epistatic effect size (user-defined, zero by default); αj is the effect size for jth covariate X.Output files are in the formats as the direct inputs for the following tools: EMMAX14, Blossoc4, Plink (.ped file), QTDT15, TASSEL16, GenABEL17 (see Table 1).
Applying one of the options shown below controls the output format. Each output format has a special suffix type, which defines the file format. These output formats are concordant to those used in phenosim.
cophesim is freely available for download from the following link: https://bitbucket.org/izhbannikov/cophesim. Requirements: Python v2.7.10 and newer, plinkio v0.9.6, R v3.2.4 and newer, Plink v1.07, - in order to run the examples. The user manual is provided in a separate file “cophesim.pdf” located in the program directory and is also available as Supplementary File 1.
Below we present an example that shows simulation of genetic data and then simulation of three different phenotypic traits. Other examples and installation instructions are provided at the program website and also in the user manual. Refer to the user manual for description of input parameters.
#-------------------------------------------Example begins---------------------------------------#
#Step 1: genetic data simulation:
plink --simulate-ncases 5000 --simulate-ncontrols 5000 --simulate wgas.sim --out sim.plink --make-bed
#Step 2: Convert .bed to .ped:
plink --bfile sim.plink --recode --out sim.plink
#Step3: phenotype simulation from previously made genetic data:
python cophesim.py -i sim.plink -o testout -itype plink -otype plink -c -ce effects.txt -s -gomp
#-------------------------------------------End of example---------------------------------------#In this example, we first (Step 1) simulate genetic data using Plink. We simulate N.cases = N.control = 5,000 cases and controls and 1,000 SNPs (defined in wgas.sim file, refer to the Plink website to see documentation for this type of file). Then (Step 2) we convert a binary sim.plink.bed file to sim.plink.ped (option --recode in Plink). This step is not required since cophesim can handle binary Plink files (.bed, .bim, .fam), but we provide this step in order to show the ability of the program to deal with Plink PED format. Finally (Step 3), we simulate dichotomous (by default), continuous (option -c) and survival (option -s) traits from previously simulated data stored in files sim.plink.ped and sim.plink.map. Note that we simulate survival trait with Gompertz hazard function (option -gomp); effect sizes for causal variants are provided in file effects.txt (to include this file we use option -ce).
We provide Receiver-Operating Characteristic (ROC) curves (Figure 2) constructed from association tests performed on a simulated dataset. Simulation and association testing were performed with Plink suite. The following parameters were used: N = 10,000 individuals, N.snp.c = 100 causal, with total N.snp = 1,000 variants. Causal variants were labeled with ‘1’ and the other (neutral) variants were labeled with ‘0’. These labels are then used later as true identifiers during calculation of TPR (true positive rate) and FPR (false positive rate). Dichotomous, continuous and survival phenotypic traits were simulated with cophesim. Then association tests were performed with Plink for dichotomous and continuous traits (using Plink flags –logistic and -regression, respectively). Association tests for survival trait were performed with the R package GenABEL. Then calculated p-values provided by association tests for each variant were compared to the significance threshold. Those variants passed the threshold were recognized as causal and associated with simulated phenotype. These classification results later were compared to the true identifiers (defined above) in order to obtain TPR and FPR. For all these tests, we varied the significance threshold from 0 to 1 with the increment of 0.001.

TPR: True Positive Ratio, FPR: False Positive Ratio. These results were calculated for dichotomous, continuous and survival traits. The dashed, 45 degrees line represents random guessing.
The R code to construct ROC curves is provided in the file “roc.R”. This file is attached to this computer note and also in the data repository: https://bitbucket.org/izhbannikov/cophesim_data/ROC/roc.R
In this work we presented the cophesim for phenotype simulation from genetic data obtained either from simulation or real data collecting. cophesim makes it possible to simulate various demographic models under user-defined scenarios.
Tool and source code available from: https://bitbucket.org/izhbannikov/cophesim
Archived source code as at time of publication: doi:10.5281/zenodo.81019518
License: MIT
The example script and output files for the software are available at: https://doi.org/10.5281/zenodo.80409019.
To test the cophesim we provided a repository “cophesim_data”: https://bitbucket.org/izhbannikov/cophesim_data. Download or clone this repository to be able to run tests.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)