Keywords
genome editing, DNA excision, sRNA, transposase, chromatin remodelling,
genome editing, DNA excision, sRNA, transposase, chromatin remodelling,
Paramecium, like other ciliates, contains both a large, somatic, polyploid macronucleus (MAC), and a relatively tiny, gametic, diploid micronucleus (MIC), within the same cell. During sexual or asexual “reproduction” (conjugation or autogamy), a copy of Paramecium’s micronucleus develops into a new macronucleus, and at the same time the micronuclear genome it contains is transformed into a new macronuclear genome by extensive, targeted DNA deletion. Concomitant with DNA deletion, the developing MAC genome is amplified from 2N to ~800N. In Paramecium, the best studied germline DNA sequences destined to be deleted, known as internally eliminated sequences (IESs), are interspersed among the macronuclear-destined sequences (MDSs) (Figure 1). Characterized Paramecium IESs are typically short (> 90% are < 150 bp), TA-flanked, and unique (∼45,000 IESs per Paramecium tetraurelia MIC genome1). These IESs are recognized and generally precisely removed by the PiggyMac transposase complex2. For some IESs, development-specific small RNAs (sRNAs) are required for the recognition and efficient removal of all their copies3.
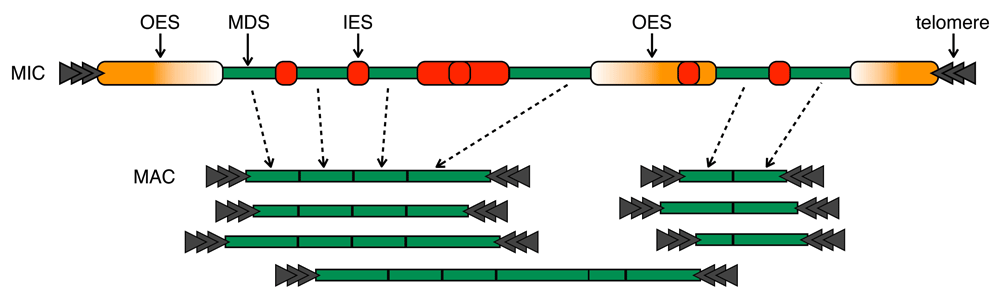
A representative micronuclear chromosome is transformed into macronuclear chromosomes via precise excision of internally eliminated sequences (IESs) and imprecise elimination of other eliminated sequences (OESs). Macronuclear-destined sequences (MDSs) inbetween are joined and capped with new telomeres to give rise to the final macronuclear chromosomes. When DNA is imprecisely eliminated, the telomere-capped ends of the macronuclear chromosomes occur at different sites. In some cases, this process also produces alternative macronuclear chromosome forms. Note that the distinction between OESs and IESs, though useful for the purposes of description and analysis here, is an artificial one, and that there may only be one main class of eliminated DNA in Paramecium (see later in Discussion).
Paramecium’s current annotated IESs comprise just a small fraction (∼13%) of all the micronuclear-specific DNA that is eliminated during development1. Though much of the remaining eliminated DNA has been assembled during IES annotation1, it has yet to be assembled as a complete micronuclear genome, and comparatively little is known about it. For the sake of explanation, and because some of this DNA may reside at chromosome ends, and hence may not be internally eliminated, we classify this DNA as “other eliminated sequences” (OESs; see Figure 1).
Developmental elimination of the few OESs investigated in detail4–6 is associated with two possible outcomes: either breakage of the MIC chromosomes into smaller MAC chromosomes, accompanied by end capping with telomeres, or fusion of the regions flanking the removed region5–7. The lengths of studied OESs are on the order of kilobases to tens of kilobases4–6. The boundaries of MAC chromosome telomere addition sites occur at similar locations to the boundaries of the internally removed sequences6. In contrast to classical IESs, the cut sites of these internally removed sequences are imprecise6. During chromosome breakage, the exact locations of the telomere addition sites vary over a span of at least hundreds of base pairs, but whether this is due to resectioning of DNA before telomere addition or due to initial imprecise DNA cleavage, or both, is unknown6.
An important question has therefore been whether the mechanisms underlying imprecise DNA elimination are distinct from those employed in precise IES excision, as previously proposed6, or whether some of the same developmental DNA deletion pathway is shared, as previously proposed4. Suggesting the latter may be true, substantial retention of OESs occurs after silencing the gene encoding PiggyMac (PGM)1, and OESs and transposons embedded within them are typically retained when gene silencing leads to IES retention3,8–12.
In ciliates, small RNAs (> 21 nt and < 32 nt) involved in MAC genome development have proposed roles either in DNA targeting and excision (as in Tetrahymena and Paramecium13–16) or in marking/protecting DNA for retention (as in Oxytricha17,18). Paramecium tetraurelia has two classes of sRNAs involved in DNA elimination: scnRNAs and iesRNAs3,13. scnRNAs are produced in gametic MICs during micronuclear meiosis, early during sexual development. Newly formed scnRNAs are transported to the existing, old MAC where MDS-matching scnRNAs are removed (i.e. removed by “RNA scanning”, as originally proposed in the ciliate Tetrahymena19), after which the non-MDS-matching scnRNAs (including IES-matching scnRNAs) are transported to the developing new MAC where they target DNA deletion3,13. Paramecium’s scnRNAs have been proposed to bind to longer RNA transcripts, rather than directly to DNA, in both the old and new MAC12,20. iesRNAs are produced in the developing MAC, where, in principle, they can immediately target unexcised IESs, and their production peaks later during development3. iesRNAs are hypothesized to be produced from transcripts of excised IESs3.
scnRNAs and RNA scanning were discovered in the ciliate Tetrahymena (belonging to class Oligohymenophorea, like Paramecium)14,16,21. Other than the use of scnRNAs to target excision, Tetrahymena’s macronuclear genome development resembles that of Paramecium in a number of other key ways, most notably in employing a domesticated PiggyBac in DNA elimination22, and that both species have two phases of development-specific sRNA production16. There are also important differences between Paramecium and Tetrahymena genome development, in particular, unlike Paramecium’s classical short, precisely excised, single copy IESs, Tetrahymena’s conventional IESs tend to be longer, imprecisely excised, multicopy elements23,24. In contrast to Paramecium, which produces scnRNAs and iesRNAs with distinct Dicer-like proteins (Dcl2/3 for scnRNAs13; Dcl5 for iesRNAs3), Tetrahymena produces two types of scnRNAs (early-scnRNAs and late-scnRNAs16) with a single Dicer-like protein (Dcl1)25,26. These scnRNAs are differentiated by the Piwi proteins (Twi1 and Twi11) processing Dcl1-generated dsRNA precursors16. At present, it is not known how the development-specific sRNAs of Paramecium and Tetrahymena guide the domesticated PiggyBacs to their DNA targets, nor what role proteins implicated in their genome editing have in mediating the intervening interactions.
In the past, the effect on the excision of Paramecium IESs following microinjection of IES DNA copies into the old MAC during autogamy was investigated for a small set of IESs. IESs were classified either as maternally controlled (i.e. governed by the old or maternal MAC) when the DNA injection led to IES retention in the new MAC, and non-maternally controlled when there was no observable IES retention27,28. In principle, since DCL2/3 co-silencing stops transmission of epigenetic information from the old macronucleus to the new one by eliminating the scnRNAs bearing this information, we previously classified IESs affected by DCL2/3 silencing as epigenetically controlled, and those that were not, as non-epigenetically controlled3,15. This classification is a simplification, since there is no clear boundary between epigenetically and non-epigenetically controlled IESs (and between maternally and non-maternally controlled IESs), and because it may be difficult to detect low levels of epigenetic control. After DCL2/3 silencing, some non-maternally controlled IESs are weakly retained, below the detection level of conventional PCR analyses3, and hence are weakly epigenetically controlled. IESs affected by DCL5 silencing do not correlate well with those affected by DCL2/3 silencing, suggesting that a substantial amount of Paramecium's sRNA-dependent IES excision is not epigenetically controlled3,15.
In the last few years, additional proteins influencing IES removal in Paramecium have been reported, including homologs of proteins with known functions in other eukaryotes, such as those involved in histone modification (PtCAF1, Ezl1) and RNA transcription (TFIIS4)10–12 (see also Table 1 and Table 2). From these studies, following existing models in Tetrahymena29,30, this has led to the proposal that scnRNAs in Paramecium direct certain chromatin modifications that may facilitate DNA excision10,11. Proteins involved in IES removal with no readily detectable homologs in other eukaryotes have also been investigated8. Other than these proteins, two closely related protein paralogs, Nowa1 and Nowa2 (Nowa1/2), were previously implicated in IES elimination and proposed to be involved in scnRNA-mediated, trans-nuclear crosstalk31. Identification and establishment of the relationships between all these proteins and the development-specific sRNAs is necessary to determine how genome editing functions in Paramecium, and the main objective of the research presented here.
Effects of knockdowns as described in this and existing studies. See Results for a description of the functional group assignment.
| Knockdown | IES excision affected | Long IESs strongly affected | Transposon excision affected | OES elimination affected | Effect on scnRNAs | Effect on iesRNAs | Effect upon H3K27me3 and H3K9me3 | Functional group |
|---|---|---|---|---|---|---|---|---|
| PGM | All known1 | Yes | Yes1 | Yes | Reduction3 | Reduction3 | None11 | DNA excision |
| DCL2/3 | scnRNA- and indirectly, some iesRNA- dependent3 | Yes3 | Yes3 | Yes12 | Elimination3 | Reduction3 | Reduction in old and new MAC11 | RNA-associated |
| DCL5 | iesRNA- dependent3 | No3 | Very little3 | Very little | None3 | Elimination3 | None11 | RNA-associated |
| DCL2/3/5 | scnRNA- and iesRNA- dependent | Yes | Yes | Yes | Elimination | Elimination | Not determined | RNA-associated |
| NOWA1/2 | scnRNA- and iesRNA- dependent | Yes | Yes31 | Yes | Nonremoval of MAC genome- matching scnRNAs | Reduction | Reduction in old and new MAC3 | RNA-associated |
| TFIIS4 | scnRNA- and iesRNA- dependent12 | Yes12 | Yes12 | Yes12 | Little reduction, if any12 | Elimination12 | Not determined12 | RNA-associated |
| EZL1 | scnRNA-, iesRNA- dependent and other11 | Yes11 | Yes11 | Yes11 | Not determined11 | Not determined11 | Reduction in old and new MAC11 | Histone modification |
| PTCAF1 | scnRNA-, iesRNA- dependent and other10 | Yes | Yes10 | Yes | Nonremoval of MAC genome- matching scnRNAs10 | Reduction10 | Reduction in old and new MAC10 | Histone modification |
| PDSG2 | scnRNA- and iesRNA- dependent | Yes | Yes8 | Yes | Little reduction, if any8 | Elimination8 | Increase in new MAC8 |
Proteins are those considered in this study. Timing of expression during development is gauged from microarray data33. Localization is that determined from GFP fusion proteins. Putative function indicates the clearest known function.
| Protein | Timing of expression during development | Localization | Putative function |
|---|---|---|---|
| Pgm1,2 | Early2 | New MAC2 | DNA elimination |
| Dcl2/Dcl313 | Early13 | MIC13 | scnRNA production |
| Dcl53 | Late3 | New MAC3 | iesRNA production |
| Nowa1/231 | Early to Late31 | Old and new MAC31 | Linking Piwi-bound sRNAs to RNA transcripts |
| TFIIS412 | Late12 | New MAC12 | Transcription of IESs in developing MAC |
| Ezl111 | Early11 | MIC, old and new MAC11 | Trimethylation of histone H3K9 and H3K27 |
| PtCAF110 | Early10 | Old and new MAC10 | Histone H4 binding; Trimethylation of histone H3K9 and H3K27 |
| Pdsg28 | Late8 | New MAC8 | Unknown |
In this paper, highlighting the utility of DNA retention analyses in discovering associations between genome editing proteins, we present what we have learnt about the role of Nowa1/2 proteins and their relation to other Paramecium genome editing components. We report strong correlations between the IESs affected by NOWA1/2 co-silencing (NOWA1/2-KD; KD=knockdown), DCL2/3/5 triple silencing (DCL2/3/5-KD) and TFIIS4 silencing (TFIIS4-KD), suggesting that Dcl2/3/5, Nowa1/2 and TFIIS4 are all components of an sRNA-guided DNA excision subsystem. Strong correlations also exist between PTCAF1-KD and EZL1-KD IES retention, whereas the correlations between the IES retention of these gene knockdowns and those of RNA-related genes are somewhat weaker. We also observe more IES retention due to the silencing of chromatin modifying genes (PTCAF1 and EZL1) than due to depletion of most IES-matching sRNAs by DCL2/3/5-KD, suggesting that chromatin modification due to PtCaf1 and Ezl1 also influences genome editing in an sRNA-independent manner. The correlations in OES retention following gene knockdown suggest most OESs are effectively the longest IESs.
All experiments were carried out with Paramecium tetraurelia stock 51, which may be obtained from the ATCC as ATCC30567. Cells were grown in a wheat grass powder (WGP; Pines International, Lawrence, KS) infusion medium bacterized the previous day with Klebsiella pneumoniae supplemented with 0.8 mg/l β-sitosterol. Cultivation and autogamy were carried out at 27°C as previously described28.
Gene silencing (knockdown) was carried out by RNA interference by feeding of Paramecium cells with E. coli expressing doubled-stranded RNA from a portion of the target gene cloned into the L4440 vector, as previously described3,8,10,31. Sequence regions used for each of the genes silenced in DCL2/3/5-KD were as previously described3,13. Silencing of PTCAF1 and ND7 were as previously described10,31.
Three knockdowns of NOWA1/2 were performed; where necessary we distinguish between these knockdowns (i.e. NOWA1/2_a-KD, NOWA1/2_b-KD, NOWA1/2_c-KD). Macronuclear DNA from postautogamous cells from the first and third knockdowns was used to produce Illumina paired-end sequence libraries. Illumina sRNA libraries were produced from NOWA1/2_b-KD at the same time as those produced for sRNAs from DCL2/3-KD, DCL5-KD and control cells - the latter knockdowns were described in "Experiment 1" of 3.
The following controls were used in this manuscript:
Control 1 = feeding with an empty vector (EV) construct. This was the control for DCL2/3/5-KD and PDSG2-KD;
Control 2 = ND7-KD. This was the control for PGM-KD1,
Control 3 = ND7-KD. This was the control for an unpublished experiment (Singh et al., in preparation);
Control 4 = ND7-KD. This was the control for EZL1-KD11.
ND7 is a trichocyst discharge gene which is not expected to affect IES retention32.
Tests of survival of sexual progeny cells were performed by isolating and refeeding 30 individual cells. NOWA1/2 knockdown is generally lethal, i.e., typically all cells die within at most the time taken for 3 divisions of control cells following autogamy; for DCL2/3/5-KD 90% of cells were dead 3 days after autogamy; for PDSG2-KD after 3 days 50% of cells were dead and 47% appeared to be “sick” (i.e. dividing slower than usual); for PTCAF1-KD after 3 days 80% of cells were dead and 17% were sick.
Morphological characteristics of MAC development were used to assign the developmental stages of the control and knockdown samples: "Early", "Middle" and "Late", correspond to 50% of cells with fragmented MACs; 100% of cells with fragmented MACs; and 100% cells with fragmented MACs and a new MAC; Figure S3A,C). Note that because Paramecium cells are not completely synchronous during induction of autogamy33 and because the underlying assessments are somewhat subjective, due caution should be exercised when comparing developmentally staged analyses between gene knockdowns.
To assess the effectiveness of DCL2/3/5-KD, northern blotting was performed as in 3. The DCL2 probe was from gene position 857-1810 (ParameciumDB: GSPATG00003051001); the DCL3 probe was from gene position 518-1354 (ParameciumDB: GSPATG00027456001); the DCL5 probe was from gene position 2074-2461 (ParameciumDB: GSPATG00012932001). For PTCAF1-KD and control (ND7-KD) cells, northern blotting was performed as in10,31.
A portion of Nowa1's C-terminal domain (amino acids 744-1024) fused to an N-terminal Strep-Tag was expressed in E. coli BL21-CodonPlus(DE3)-RIL cells (formerly Stratagene, currently Agilent, USA). The protein was column purified, followed by removal of the tag by HRV 3C protease cleavage. Immunization of a rabbit with this protein was performed by Eurogentec, Belgium, following their 28-day polyclonal antibody protocol, then affinity purified using the matrix bound C-terminal domain portion of Nowa1, before performing western blotting using a 1:500 antibody dilution (Figure S3B). As a loading control, western blotting was performed using 1:5000 dilution of monoclonal anti-alpha-tubulin (Sigma T8203 lot 111M4828) and 2% BSA for blocking.
For small RNA electrophoretic analysis, total RNA samples (5.0 μg) were 5'-end-labelled with [γ-32P] ATP (5000 Ci/mmol, Amersham) by the exchange reaction of T4 polynucleotide kinase (Fermentas), then denatured and run on a 15% polyacrylamide, 7 M urea gel.
Synthetic Nowa1 codon adjusted for E. coli translation (GENART synthesis) was cloned into the pMAL-c2x vector and expressed in Rosetta2(DE3)p-LysS (formerly Novagen, currently Merck) cells. Protein translation was performed at 37°C; cells were supplied with 1 mM IPTG and 0.2% glucose. Protein was purified according to the pMAL™ Protein Fusion and Purification System (NEB e8200s). EMSA was performed with the LightShift® Chemiluminescent RNA EMSA Kit (Thermo scientific, 201558) using 6.25 μM of RNA or DNA, 4 μg of MBP, and 12 μg of MBP-Nowa1.
Macronuclear DNA was isolated from post-autogamous cells as previously described1. For PCR analyses of IES retention, DNA was extracted from approximately 800 cells with a Nucleospin Tissue kit (Macherey-Nagel).
Total RNA was extracted from 400 ml of culture per the TRI Reagent BD protocol (Sigma) and was resuspended in RNase free water. A MirVana miRNA isolation kit (Ambion) was used for sRNA enrichment.
MAC genomic DNA and small RNA libraries were produced and sequenced per standard Illumina protocols. Paired-end Illumina DNA-seq libraries were constructed and sequenced on either the Illumina GAIIX, HiSeq 2000 or HiSeq 3000 sequencers.
Sequence data corresponding to the MAC DNA from NOWA1/2_a-KD can be obtained from the GenBank Sequence Read Archive (SRA) under the accession SRR646462. MAC DNA sequence data for NOWA1/2_c-KD, DCL2/3/5-KD, PTCAF1-KD, PDSG2-KD, ND7-KD and control 1 (empty vector) is available from the European Nucleotide Archive, respectively under the accession numbers ERS1033672, ERS1033670, ERS1033674, ERS1033673, ERS1033676, ERS1033671. sRNA sequence data for NOWA1/2_b-KD and empty vector control can be obtained from the European Nucleotide Archive accessions ERS1455004, ERS1455005, ERS1455006, ERS1455007, ERS1455008, ERS1455009, and for DCL2/3/5-KD, from ERS1455358, ERS1455359, ERS1455360.
PCR conditions and primers used to test IES retention following NOWA1/2_a-KD were the same as those in 3.
The following reference genomes for Paramecium tetraurelia stock 51, obtained from ParameciumDB34, were used in the IES analyses and read mapping:
MAC: http://paramecium.cgm.cnrs-gif.fr/download/fasta/assemblies/ptetraurelia_mac_51.fa
MAC+IES: http://paramecium.cgm.cnrs-gif.fr/download/fasta/assemblies/ptetraurelia_mac_51_with_ies.fa (md5 checksums: dbcc54fb2987c8f60f8e765db7ed274c and 3e5b3fa65ebfaa484566a1ffddf20239, respectively).
IES retention scores (IRSs) were determined by ParTIES35. For each IES, ParTIES counts mapped reads with unexcised IESs (IES+) and excised IESs (IES-) to determine an IES retention score, IRS = IES+ ÷ (IES+ + IES-). A table of all the IES retention scores used in this manuscript is included as Dataset 1 (see Data and software availability). IES retention scores from previous knockdown experiments, i.e. PGM-KD1, TFIIS4-KD12, EZL1-KD (EZL1-2 silencing fragment)11, DCL2/3-KD3 and DCL5-KD3, were downloaded from ParameciumDB34. Unless otherwise indicated, NOWA1/2-KD IRSs are for NOWA1/2_a-KD.
To generate the IES and OES score regression matrices, shown in Figures 3A–C, Figure 4E and Figures S1A–B, we provide the program “after_ParTIES.py” (see Data and software availability), which can be run on Dataset 1 and Dataset 2 (IES and OES retention scores, respectively). This was tested using Python3.6 and relevant modules for scientific computing from the Anaconda software distribution from Continuum Analytics (version: Anaconda3-4.3.1).
Three regressions are graphically shown for IRS pairs: an ordinary least squares (OLS) regression, a LOWESS regression (non-parametric) and an orthogonal distance regression (ODR) with a linear function initialized by the slope and intercept from the OLS. OLS and ODR used standard SciPy functions. LOWESS regressions used the Python statsmodels library. ODR was performed since errors in both the "dependent" and "independent" regression variables are expected (both IRSs), not just the dependent variable as assumed by OLS. The combined effect of background IES retention and systematic IES retention errors on the regressions, and the effect of subtracting the mean of multiple control IES retention scores is shown in Figure S1A. For each pair of knockdown IES retention scores, Spearman’s rank correlation coefficient (rs) was calculated instead of Pearson’s correlation coefficient to mitigate the effect of outliers. Reproducibility of IES retention scores between experiments is demonstrated by the strongly correlated scores of different NOWA1/2 silencing experiments (Figure S1B).
Two-sided p-values, calculated with SciPy’s spearmanr function, were used to test the null hypothesis of no correlation (for p ≤ α/m ≤ 0.01/45; i.e. with a Bonferroni correction for multiple hypothesis testing; uncorrected p-values are given in Supplementary Data S1–4), which was rejected in all the displayed correlations in the main figures, except where indicated by a circumflex accent. To facilitate assessment of the statistical significance of differences between rs values, tables containing the two-tailed p-values are provided in Supplementary Data S5-7. p-values were calculated using methods from the R statistical package cocor version 1.1-336 that employ Fisher’s r-to-Z transformation (which are also applicable to rs37). Appropriate methods for calculating test statistics for correlations with independent groups or dependent groups with either overlapping or non-overlapping variables were used from cocor (noted in the supplementary data files). All the comparisons of rs differences described in the text are statistically significant at α=0.01 with a Bonferroni correction for multiple hypothesis testing.
Genomic regions that are not part of the current reference Paramecium MAC+IES assembly were previously assembled1, but we decided to use an alternative, metagenome assembler to attempt to produce a better assembly, and to minimize bacterial contamination. Reads from PGM-KD1 not mapping to the current Paramecium tetraurelia strain 51 MAC genome and 51 MAC genome with IESs (bwa38 version 0.7.12-r1039; default parameters) were assembled with IDBA-UD39 version 1.0.9 (default parameters). To remove bacterial contaminants assembled during the assembly, only scaffolds with GC < 40% were selected (bacterial scaffolds peak around 58% GC; Figure S5A). This procedure resulted in a ∼20 Mb assembly consisting of 7266 scaffolds. As a small fraction of the P. tetraurelia MAC genome is currently missing from the reference assembly, this fraction was present in the ∼20 Mb assembly. To exclude the missing MAC genome regions, empty vector control (control 1) reads were mapped to the 7266 scaffolds with HISAT240 version 2.0.4 (parameters: “-X 600 --pen-noncansplice 0 --mp 24,8 --max-intronlen 1000), and then, using a custom Python script, only those regions with little or no sequence coverage (≤ 2.0×; compared to a mean MAC coverage of ∼100×) were extracted (Figure S5C shows an illustrative scaffold from which OESs were extracted). Coverage was smoothed using a bi-directional average of two exponentially weighted moving averages (EWMA) with a 200 bp span. This procedure extracted 4799 sequence regions (12.3 Mb).
To minimize the effects of potential genome assembly chaff (short assembled regions, esp. those that might arise from sequence errors), analyses were continued with only the low control sequence coverage regions extracted from scaffolds longer than 300 bp and with a PGM-KD sequence coverage of between 3× and 70× (Figure S5B). These 3882 regions, obtained from 5236 scaffolds, corresponding to 11.8 Mb of sequence data, are classified as putative OESs (Supplementary Data S8). Note that OES boundaries are a first approximation, since imprecise DNA elimination generates variable boundaries (as illustrated in Figure 1 and shown in Figure S5C), and many OESs situated at the ends of the scaffolds are likely incomplete due to the limitations of genome assembly (esp. since the assembly may end when untraversable repetitive regions are encountered).
For each knockdown, to quantify the effects of gene knockdown on OES retention, reads were mapped to the OESs (identified in the previous methods section) with HISAT240 version 2.0.4 (parameters: “-X 600 --pen-noncansplice 0 --mp 24,8 --max-intronlen 1000) before determining coverage, and normalized to the median coverage of the knockdown reads mapped with HISAT2 to the Paramecium tetraurelia strain 51 MAC genome plus IES assembly, i.e. OES retention score=OEScoverage/median(MAC-with-IES-genomecoverage). OES retention scores may not be as accurate as IES retention scores, both because their denominators use a measure of global, rather than local, sequence coverage and because only reads that map within OES boundaries are counted. In the latter case, read mapping may fail to detect short matches to an OES when only a short part of an OES/MDS-derived read matches, and so the shorter the OES, the greater the underestimation of its retention. Shorter OESs are more weakly retained in most knockdowns affecting genome reorganization (Figure S5D) in Paramecium, but at least some of this effect may be due to boundary-related underestimation of retention. In future, when a complete Paramecium MIC genome is assembled, the relationship between OES length and retention can be fully assessed. OES retention estimates are provided in Dataset 2. Control OES retention was subtracted from the knockdown OES retention for each OES used in the regression analyses.
sRNAs were mapped and histograms of their genomic distribution were produced as previously3 with the addition of OESs.
Sequence logos were created with WebLogo41, version 3.3, with redundant sRNAs and base frequencies A=U=0.4 and C=G=0.1.
Previously, each newly silenced Paramecium gene involved in IES excision appeared to generate a different distribution of IES retention3,11,12. From additional knockdowns we have subsequently performed, it can now be seen that the IES retention distributions for NOWA1/2 knockdown, TFIIS4 knockdown and DCL2/3/5 triple knockdown are very similar (Figure 2A; see Methods for the IES retention score calculation). Given the role of TFIIS4 and Dcl2/3/5 in producing and processing of RNAs, and the suggested role of Nowa1 in RNA interactions (see “Nowa1/2 is involved in RNA scanning, may mediate scnRNA- and iesRNA-targeted DNA cutting and excision” later in the results), we classify these proteins as "RNA-associated" proteins involved in Paramecium MAC genome development. The IES retention score distributions of the EZL1 knockdown and PTCAF1 knockdown also have similar forms, though the effect of the former knockdown is slightly stronger (Figure 2B). Since both Ezl1 and PtCAF1 affect histone modifications10,11, we classify these proteins as those involved in "histone modification".
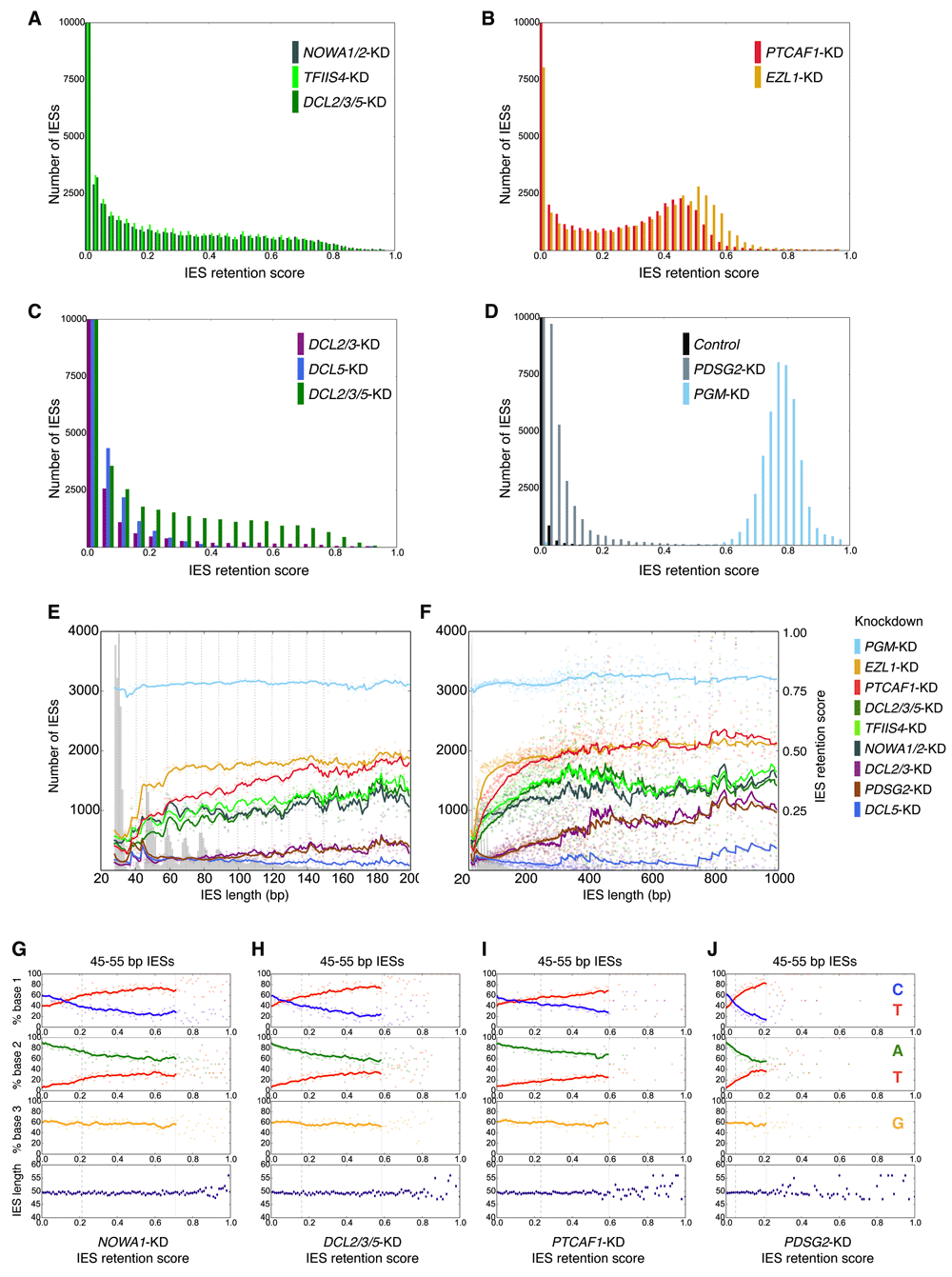
(A–D) For the sake of clarity, histogram bars with more than 10000 IESs have been truncated. The number of IESs binned in the left-most bars are as follows: (A) NOWA1/2-KD - 17468, TFIIS4-KD - 14152, DCL2/3/5-KD 18915 (Figure S3B and S3D provide Western and Northern blots, for NOWA1/2-KD and DCL2/3/5-KD, respectively); (B) PTCAF1-KD - 12614, EZL1-KD - 8047; (C) DCL2/3-KD - 37410, DCL5-KD - 35019, DCL2/3/5-KD - 22136; (D) Control - 43294; PDSG2-KD - 20730. Note that, as judged by survival tests in comparison to those previously published 8, PDSG2-KD may have been of reduced efficiency. (E,F) IES retention scores (scales on right y-axes) vs. IES length over shorter (E) (≤ 200 bp) and longer (F) (≤ 1000 bp) IES length scales. The IES length distribution is shown in grey in the background (scale on left y-axes). Lines are the exponentially-weighted mean averages (EWMA) with spans of 5 (E) or 50 (F) bp. (G–H) Mean base frequencies of positions 1-3 after the TA repeat relative are plotted relative to IES retention score for IESs from the third (45-55 bp) length peak (only the highest frequency bases are shown — see (H) for the base color scheme; similar trends are visible for ~10 bp windows surrounding longer IES length peaks). IES length is restricted to control for IES end base frequencies variation associated with large scale (26-1000 bp) IES length variation. EWMA lines for spans of 10 data points (intervals of 0.01) are plotted for data within 2 standard deviations (dotted vertical lines) of the mean retention score (dashed vertical line). Mean IES lengths (bp; counting only one of the two TA pointers) vs. IES retention score are potted in the bottom subgraphs (navy diamonds). See Figure S2 for end base frequency graphs of EZL1-KD and TFIIS4-KD and for end base frequency graphs of shorter IESs (26-36 bp). Equivalent graphs for DCL2/3-KD, DCL5-KD and PGM-KD can be seen in 15.
As we previously reported, the effects of either DCL2/3 knockdown or DCL5 knockdown on IES retention alone are quite modest3,15. Even when their IES retention scores are summed, it is apparent that the knockdowns of DCL2/3 and DCL5 influence considerably fewer IESs, and to a lesser degree than the PGM knockdown. For DCL2/3/5, NOWA1/2 and TFIIS4, given that the gene knockdowns are efficient (as judged by depletion of their mRNAs or proteins; Figure S3B,D and12), we think the similarity of their IES retention score distributions indicates that the upper bounds of IES retention for these knockdowns have been approached. Consequently, these knockdowns may indicate most of the Paramecium IESs requiring scnRNAs and iesRNAs for their excision.
Though an interesting idea, we find no evidence of a proposed missing class of sRNAs11,12 responsible for targeting and excision of additional IESs. Instead, we observe that after DCL2/3/5-KD the levels of IES-matching sRNAs are negligible (Figure 5C). We therefore infer that most IES excision in Paramecium does not require IES-targeting sRNAs. This is consistent with past studies reporting that most tested IESs were non-maternally controlled27,28, i.e. that scnRNAs alone are insufficient for most IES excision3,15. On the other hand, from IES retention following DCL2/3/5-KD we infer that many IESs require some scnRNAs or iesRNAs to cleanly remove all their copies (the exact number of affected IESs depending on the chosen retention score threshold, e.g. 19043 IESs are affected by DCL2/3/5-KD above a threshold of 0.1 retention).
An important observation from the DCL2/3/5 knockdown is the considerably stronger IES retention compared to the separate DCL2/3 and DCL5 knockdowns (Figure 2C); for example, at an IES retention score > 0.1, for DCL2/3/5-KD 19043 IESs are affected vs. 4815 IESs for DCL2/3-KD, and 5384 IESs for DCL5-KD. This suggests some co-operation between scnRNAs and iesRNAs in IES excision. From the DCL2/3/5 triple silencing, it is clear that multiple gene co-silencing may be necessary to gauge the full contributions of individual genes to IES excision. This is a critical consideration in gene function investigations in Paramecium tetraurelia with its abundance of paralogs that arose from successive whole genome duplications42.
As we previously proposed, IES excision/recognition efficiency and the need for particular genome reorganization proteins vary as a function of IES end bases and lengths15. A trend of increasing IES retention with IES length was shown for the knockdowns of DCL2/3, TFIIS4 and EZL13,11,12,15. It can now be seen that, with the exceptions of PGM-KD and DCL5-KD, most knockdowns leading to IES retention show this trend (Figure 2E,F). A new observation with respect to IES length is that IES retention for the knockdowns of DCL2/3/5, TFIIS4 and NOWA1/2 also has an obvious 10-11 base periodicity for ~45-130 bp IESs (Figure 2E), mirroring that of the IES length peaks (which are proposed to reflect the constraints of DNA twist on transposase excision1). This signal is not apparent in knockdowns with weaker effects (DCL2/3-KD, DCL5-KD and PDSG2-KD), and only weakly visible in the knockdowns with stronger effects (PTCAF1 and EZL1). We previously also observed that the terminal IES base frequencies relative to IES length show a periodic, 10-11 bp signal (Figure 2A of 15). As in knockdowns of DCL2/3, DCL5 and PGM15, for all the new knockdowns we examined, the end base frequencies of longer IESs (e.g. 45-55 bp) vary in a similar manner with respect to their IES retention scores (Figure 2G–H; Figure S2), consistent with the idea that some IESs have a greater requirement for the products of these genes than others15.
While similar IES retention distributions suggest different gene knockdowns might affect retention of particular IESs in a similar manner, examination of pairwise correlations is necessary to clearly establish this. We therefore determined all the possible pairwise correlations between IES retention scores of different gene knockdowns (Figure 3). From this figure, it can be seen that the DCL2/3/5, NOWA1/2, TFIIS4 (group 1), and histone modification genes (group 2) not only have the most similar IES retention score distributions within each of these two groups, but also the strongest IES retention score correlations. Though not as strong as those within the DCL2/3/5, NOWA1/2, TFIIS4, and histone modification groups, positive correlations are also evident between these groups (Figure 3; e.g. Spearman’s rank correlation coefficient, rs=0.74 between DCL2/3/5-KD and PTCAF1-KD). The correlations between PGM knockdown and other gene knockdowns are generally weak — not much greater than the correlations between the control (ctrl3) and other gene knockdowns. Such weak correlations are an expected consequence of the ability of PiggyMac to excise most IES copies without scnRNAs/iesRNAs and without chromatin state changes brought about by Ezl1 and PtCAF1.
Since IES retention increases with IES length for many of the knockdowns influencing IES excision (see Figure 2E,F), we examined the retention score correlations between the abundant IESs corresponding to the first length peak (e.g. ≤ 35 bp; ∼15600 IESs; the IES length peaks can be seen in the background of Figure 2E) compared to longer IESs (e.g. ≥ 500 bp; ~380 IESs; patterns of correlations between these length intervals are intermediate to those in Figure 2E,F). For short IESs there are good correlations between all the possible pairs of IES retention scores from DCL2/3/5-KD, NOWA1/2-KD, TFIIS4-KD, PTCAF1-KD and EZL1-KD. This could be because the protein products of all the genes involved in these knockdowns are cooperating in the excision of short IESs. For the subset of short IESs, the right-skewed IES retention score distributions of these knockdowns also all resemble each other (diagonal of Figure 3B). We previously noted that DCL5-KD leads to greater retention of short IESs than DCL2/3-KD, i.e. iesRNAs are marginally more important for short IES excision than scnRNAs and short IESs are generally weakly epigenetically controlled3,15. This is reflected by the stronger correlations in IES retention score for short IESs between DCL5-KD and the knockdowns of NOWA1/2, TFIIS4, PTCAF1 and EZL1, relative to the correlations between DCL2/3-KD and the same genes.
When focusing on longer IESs (e.g., those > 500 bp) the IES retention score correlations between the knockdowns of histone-associated genes, and also those between DCL2/3/5, NOWA1/2, TFIIS4 and histone-associated genes weaken substantially. Compared to their short counterparts the retention score distributions of longer IESs also look very different, with those of DCL2/3, DCL2/3/5, NOWA1/2, TFIIS4 closer to uniform, and those of the histone modification group closer to normal. The forms of the histone modification group IES retention score distributions now also become more like that of PGM-KD. These patterns are consistent with the chromatin state changes brought about by EZL1-KD and PTCAF1-KD exerting more influence on long IESs than short ones, with a much lesser requirement for iesRNAs, and generally some requirement for scnRNAs (i.e. long IESs are generally more strongly epigenetically controlled). At longer scales, the effect of PTCAF1-KD and EZL1-KD on IES retention (e.g. at ≥ 500 bp, rs=0.58) does not correlate as well as at the shorter scale (e.g. at ≤ 35 bp, rs=0.88), likely reflecting that the products of these genes do not have exactly the same consequences on chromatin state at this scale. The decrease in IES retention score correlations between either DCL2/3-KD or DCL2/3/5-KD and histone modification groups largely reflects that iesRNAs have little influence on long IES excision.
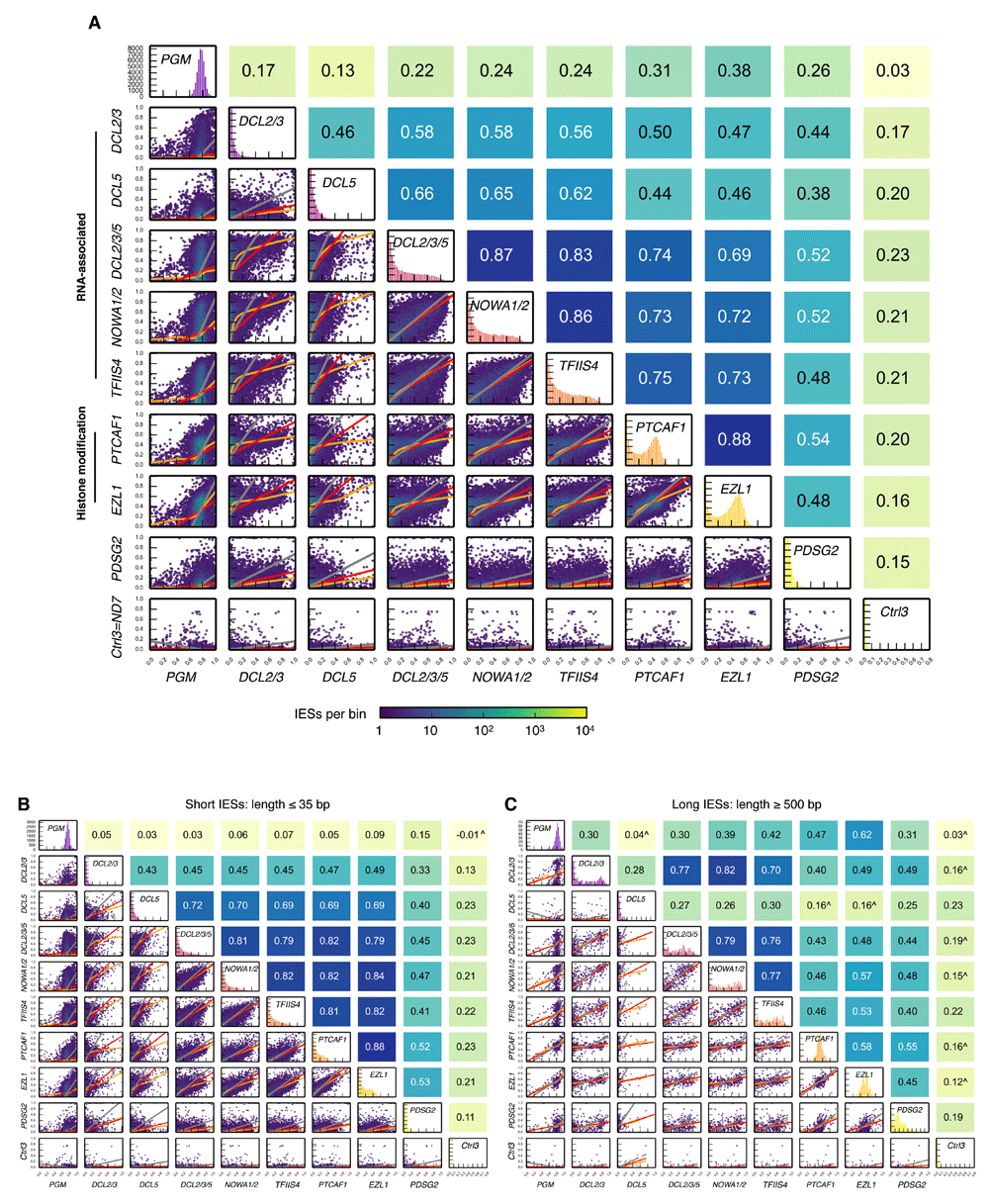
(A) For the purposes of regression, for each IES, negative control IES retention scores have been subtracted and zeroed when the subtraction yields a negative value, i.e. IRSregression=max(IRSexperiment - mean(IRScontrols), 0) (controls=controls 1-4). This calculation was performed to reduce the effects of natural, background IES retention and systematic errors in IES retention score estimation as much as possible (see Figure S1, which examines these effects on regression). Hexagonal binning of IES retention scores was used to generate the plots in the lower triangular matrix. Red lines are for OLS regression, orange lines for LOWESS, and grey lines for ODR. IES retention score distributions given along the diagonal are shown to a maximum of 4000 IESs except for PGM-KD (maximum 8500 IESs). For each plot in the lower triangular matrix (Mi,j), Spearman’s rank correlation coefficient (rs) is given in the corresponding position diagonally opposite in the upper triangular matrix (Mj,i). Note rs=0.91 for biological replicates of NOWA1/2-KD (Figure S1B). (B) as in (A) but for short IESs (≤ 35 bp). IES retention score distributions given along the diagonal are shown to a maximum of 1500 IESs except for PGM-KD (maximum 3200 IESs). (C) as in (A) but for long IESs (≥ 500 bp). IES retention score distributions given along the diagonal are shown to a maximum of 50 IESs except for PGM-KD (maximum 75 IESs).
To date, information about the effects of gene silencing upon Paramecium’s other eliminated sequences (OESs), the remaining deleted DNA currently not annotated as IESs, has typically come from Southern blotting or dot blotting, e.g. 8,10,11,13,31, though more recent studies have also begun to investigate the overall effects of gene knockdown on these regions11,12. Most gene knockdowns that lead to IES retention also lead to retention of transposons within OESs8,10–13,31. For each of the knockdowns used to investigate IES retention, we examined OES retention by mapping the knockdown reads to genomic regions which do not match MDSs and IESs from the reference P. tetraurelia assemblies that were extracted from a PGM-KD DNA assembly (see Methods for more details). Since PGM-KD leads to the most IES retention, like previous reports11,12, we assume that much of the remaining DNA eliminated from the MIC genome can be recovered from this knockdown.
Examination of the distributions of OES retention (Figure 4A–D), generated similarly to those for IES retention (Figure 2A–D), shows that while some features of OES retention resemble those of IES retention, e.g. the forms of the NOWA1/2-KD, TFIIS4-KD and DCL2/3/5-KD OES retention distributions are roughly similar, there are also important differences. OES retention due to DCL5-KD is very weak, hardly differing from that of a negative control, and DCL2/3/5-KD OES retention is weaker than that of DCL2/3-KD. Compared to the very strong IES retention following PGM-KD versus all other knockdowns (Figure 2D), PGM-KD OES retention is not as pronounced (Figure 4D). Like IESs, in part this may be because the strength of OES retention in other knockdowns is greater, but it is also possible that DNA cutting enzymes other than PiggyMac may be involved in OES elimination.
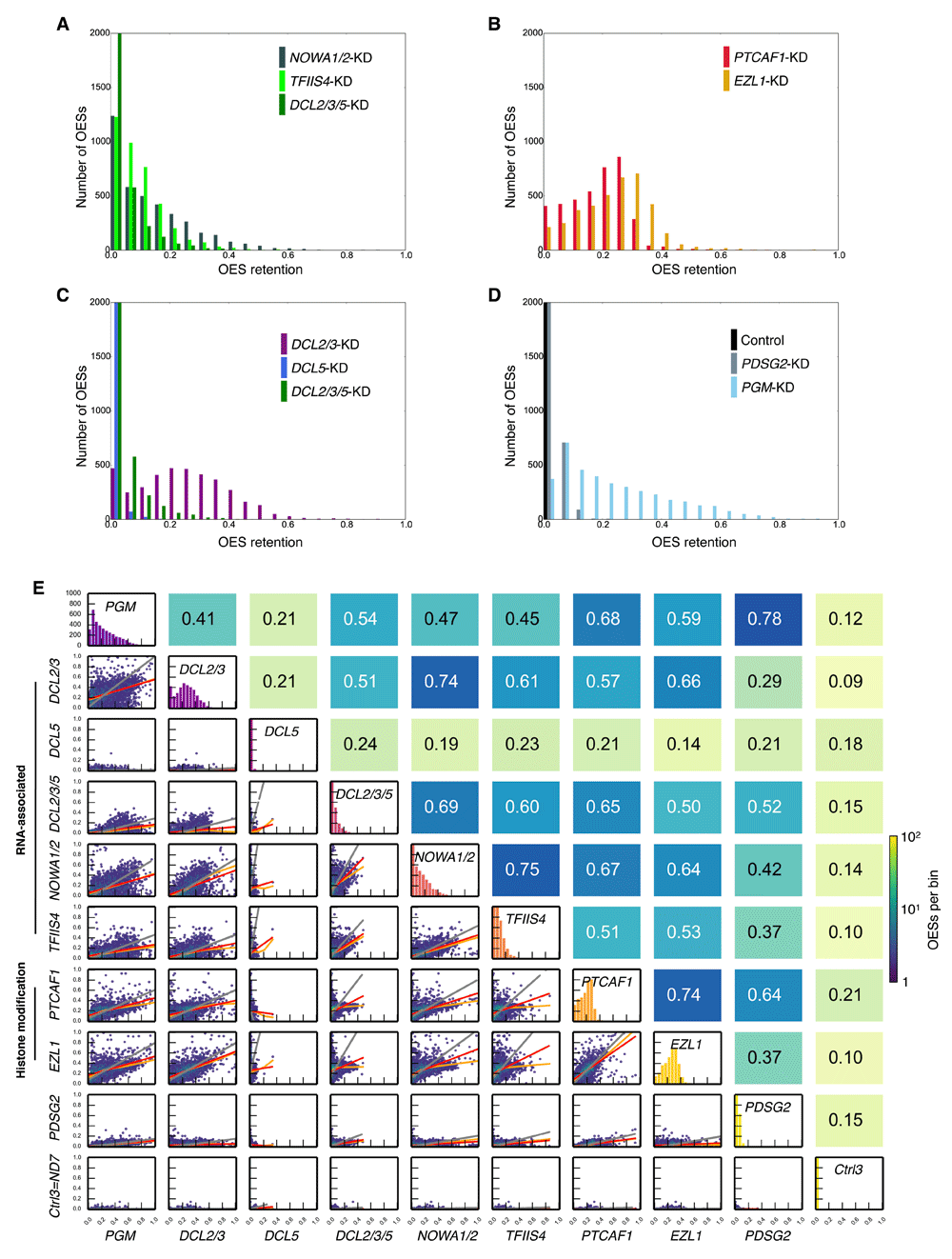
(A–D) For the sake of clarity, histogram bars with more than 2000 OESs have been truncated. The number of OESs binned in the left-most bars are as follows: (A) NOWA1/2-KD - 1238, TFIIS4-KD - 1230, DCL2/3/5-KD 2812; (C) DCL2/3-KD - 473, DCL5-KD - 3788, DCL2/3/5-KD - 2812; (D) Control - 3882; PDSG2-KD – 3062; PGM-KD – 709. (E) OES retention score correlations and regressions. For the purposes of regression, for each IES the mean of two negative control OES retention scores (control 1 and control 2) have been subtracted, and zeroed when the subtraction yields a negative value, i.e. ORSregression=max(ORSexperiment – mean(ORScontrols), 0). This calculation was performed to reduce the effects of natural, background OES retention and systematic errors in OES retention score estimation as much as possible (but note that when OES retention scores are very low, some residual background retention may lead to some correlation, e.g. between DCL5-KD and control 3). Hexagonal binning of OES retention scores was used to generate the plots in the lower triangular matrix. Red lines are for OLS regression; orange lines for LOWESS and grey lines for ODR. For each plot in the lower triangular matrix (Mi,j), Spearman’s rank correlation coefficient (rs) is given in the corresponding position diagonally opposite in the upper triangular matrix (Mj,i).
Examination of the correlations in OES retention in Figure 4E reveals a number of important effects: (i) between PGM-KD and other knockdowns, OES retention correlation is greater than general IES retention score correlations (Figure 3A); (ii) NOWA1/2-KD correlates most strongly with TFIIS4-KD and DCL2/3-KD; PTCAF1-KD and EZL1-KD correlate the best with each other, and moderately with DCL2/3-KD, DCL2/3/5-KD, TFIIS4-KD and NOWA1/2-KD; (iii) though a clear role for the Pdsg2 protein has yet to be defined, OES retention following PDSG2-KD correlates most strongly with PGM-KD; (iv) as OES length increases (e.g. > 4 kb) the PTCAF1-KD and EZL-KD OES retention scores become very narrowly distributed in contrast to those of DCL2/3-KD, NOWA1/2-KD and PGM-KD, suggesting that chromatin state changes brought about by PtCaf1 and Ezl1 may have a very general effect on OES retention. From these observations and those of the effects of previously reported analyses of the overall sequence complexity of retained OES DNA following gene silencing11,12, it follows that OES elimination mostly does not need iesRNAs, whereas scnRNAs, together with TFIIS4 and Nowa1/2 are necessary for OES elimination. The stronger correlations in OES retention scores of other knockdowns relative to those of PGM-KD suggests greater cooperation between PiggyMac, scnRNAs and other protein factors in OES elimination than in general IES excision. Overall, the patterns of OES retention correlation for the different knockdowns resemble those of retention correlations for long IESs, suggesting most OESs may be treated like long IESs.
As the Nowa1/2 proteins have previously been implicated in removal of maternally controlled IESs31, but their precise role in Paramecium genome development has not yet been established, we wished to obtain additional insight into the function of these proteins, and their relation to other molecules involved in this process. Nowa1/2 proteins contain two distinct domains: an unstructured N-terminal domain consisting of alternating blocks of ‘FRG’ repeats and ‘GGWG’ repeats, and a structured C-terminal domain which is sufficient to route Nowa1 from the maternal MAC to the zygotic nuclei31. Nowa1/2's ‘FRG’ repeats resemble the ‘RGG’ boxes present in many hnRNP and other RNA-binding proteins, raising the possibility that they could facilitate interaction between Piwi-bound sRNAs and nascent transcripts on the old and the new MAC. One way Nowa1/2 may mediate these interactions is via its "GGWG" repeats, which resemble the "WG/GW" repeats found in Argonaute binding proteins43,44 (Argonaute proteins are members of the Piwi protein superfamily45). "WG/GW" repeat-containing proteins in Tetrahymena (Wag1 and CnjB) have been shown to interact with its Twi1 Piwi protein46. In principle, Nowa1/2 could interact with Paramecium’s sRNA-bound Piwi proteins in both the old and the developing new MAC, and during the internuclear transport of the sRNAs. Like PtCAF1-KD, NOWA1/2-KD also substantially decreases H3K9me3 and H3K27me3 in the old and developing new macronuclei10.
To gain more insight into the role of Nowa1/2 in Paramecium genome reorganization we analyzed the effects of NOWA1/2-KD upon IES retention and the sRNA population during macronuclear development (Figure 5). We previously showed that in control and DCL5-KD cells, MAC genome-matching scnRNAs decrease during development, consistent with RNA scanning, and that there is a progressive increase in iesRNAs3. It can now also be seen that OESs give rise mainly to 25 nt scnRNAs and, compared to IESs, relatively few iesRNAs (Figure 5A; see also Figure S3G,H).

(A) Length distribution of small RNAs from control cells and NOWA1/2-KD cells (see Methods for details of histogram construction and normalization). Vector=vector backbone only. Figure 2SA shows the cytological characterization of the cells these sRNAs were isolated from. (B) Radioactively end-labelled total RNA from NOWA1/2-KD cells and control cells (cells fed with E. coli producing dsRNAs corresponding to the plasmid L4440 with no sequence target in the Paramecium genome; the gel portion showing the control sRNAs was previously shown in Figure 2A of 3 as "Control 1; Experiment 1"). (C) sRNA length distribution for DCL2/3/5-KD; the peak at 33 nt corresponds to an unknown sRNA family (e.g. 5'-GGAUCUAUCGUAUAGUGGUUAGUACCUGAGGCU-3') that matches a handful of locations in the MAC genome, which we have also be observed in other controls and knockdowns (e.g. from 8). See Figure 2SE for cytological characterization. (D) To examine the relationship between maternal control and IES retention scores (values determined in Table S1), maternal control scores were obtained from the maximal IES retention observed in Figure 6 of 28. Linear regression lines are shown along with Pearson's r and the two-tailed p-values for hypothesis testing with a null hypothesis that the regression slope is zero. (E) Retention of IESs tested by PCR for control and NOWA1/2-KD cells. NOWA1/2-KD IES retention scores are given to the right. Additional bands indicate the presence of heterodimers of ssDNAs with excised and unexcised IESs. (F) Electrophoretic mobility shift assay with MBP (maltose-binding protein), MBP-Nowa1 (alternating lanes) and oligonucleotides, visualized on a native polyacrylamide gel.
Since it is known that smaller IESs can be nested within larger IESs47, we searched for IESs embedded within OESs. Visual inspection of DNA reads mapping to the assembled OESs from different gene knockdowns confirms the presence of shorter IESs in OESs, often with matching iesRNAs. We previously noted that, though both scnRNAs and iesRNAs are enriched at IES ends, scnRNAs overlap IES ends, whereas iesRNAs map very specifically within the limits of IESs3. Given the patterns of OES retention shown in Figure 4E, we therefore suggest that iesRNAs primarily facilitate precise IES excision, whereas scnRNAs facilitate both precise IES excision and imprecise OES elimination.
Compared to control cells, scnRNAs accumulate and iesRNAs are reduced after NOWA1/2 silencing (Figure 5A, B). scnRNA accumulation is consistent with inhibition of RNA scanning when NOWA1/2 is silenced, i.e. MDS scnRNAs are not removed. We do not think that NOWA1/2-KD affects RNA scanning by interfering with transport of scnRNA-bearing Piwis (Ptiwi01/099), as we observed no differences in localization of GFP-tagged Ptiwi09 with and without silencing NOWA1/2 (Figure S4). On the other hand, the pronounced mobility shift in an electrophoretic mobility shift assay upon addition of synthetic MBP-Nowa1 protein (Figure 5F) indicates Nowa1 may interact with RNA, RNA:DNA hybrids, and, to a lesser degree, DNA. Hence NOWA1/2-KD-induced interference in RNA scanning in the old MAC could be due to disruption of interactions normally mediated by Nowa1/2 between nucleic acids and sRNA-carrying Ptiwi proteins.
From the electrophoretic visualization of sRNAs (Figure 5B) it can be seen that iesRNAs are reduced following NOWA1/2-KD (since Figure 5A shows only proportions, it is not possible to deduce whether absolute levels of sRNAs change from this figure). A decrease in iesRNAs was also observed following DCL2/3 co-silencing, and, to a lesser degree for the independent DCL2 and DCL3 silencings3. For the lower quantity of iesRNAs in NOWA1/2-KD cells, there are two possible explanations, both of which may be valid: (i) Nowa1/2 is directly involved in iesRNA production or stability; (ii) Nowa1/2 depletion inhibits scnRNA-dependent IES excision due to the inhibition of RNA scanning, which in turn inhibits iesRNA production. Teasing apart these possibilities is a possible future experimental avenue to gain additional insight into the function of the Nowa1/2 proteins.
NOWA1/2 silencing was previously reported to lead to retention of maternally controlled IESs, and indeed maternally controlled IES retention levels (following microinjection of complementary IESs into the old MAC) correlate well with the IES retention scores (Figure 5D). However, following NOWA1/2 silencing we also observe low levels of retention of some non-maternally controlled IESs by PCR analyses and in DNA-seq data (Figure 5D, E; Table S1). As the non-maternally controlled IESs that are weakly retained following NOWA1/2-KD are also weakly retained following DCL2/3-KD, we infer that they are weakly epigenetically controlled (Table S1). Therefore, due to the sensitivity of IES retention detection in DNA-seq data, some "non-maternally controlled" IESs may actually be weakly epigenetically controlled (it is also possible that with more sensitive methods these IESs would be classified as weakly maternally controlled). From Figure 3, we infer that Nowa1/2 is not only involved in scnRNA-dependent IES excision (epigenetically controlled IES excision), but also iesRNA-dependent IES excision, since IES retention scores of NOWA1/2-KD are positively correlated with those of both DCL2/3-KD and DCL5-KD, and strongly correlated with those of DCL2/3/5-KD.
From these observations of the effects of NOWA1/2-KD, we propose that Nowa1/2's WG/GW (Argonaute-binding) repeats could mediate IES targeting by both Ptiwi01/09-bound scnRNAs (both old and new MAC) and iesRNAs bound to other Ptiwi proteins48 in the new MAC. The apparent failure to remove MAC genome-matching scnRNAs after NOWA1/2 silencing (Figure 5A) is consistent with the involvement of Nowa1/2 in RNA scanning in the old macronucleus, as expected if these proteins mediate pairing of scnRNAs with nascent maternal non-coding transcripts. In principle, Nowa1/2 proteins could interact with such transcripts via their FRG (RNA-binding RG boxes) repeats. From the correlations in Figure 4, we infer that Nowa1/2 proteins may operate predominantly in concert with scnRNAs and not iesRNAs during OES elimination.
The strong similarity of IES retention following silencing of DCL2/3/5, NOWA1/2 and TFIIS4 suggests that the protein products of these genes may work in the same sRNA-dependent DNA elimination pathway during short IES excision, and that DCL2/3, NOWA1/2 and TFIIS4 work in the predominantly scnRNA-dependent DNA pathway responsible for OES and long IES elimination. PGM-KD-induced DNA retention correlates very weakly with that of other knockdowns for short IESs, but moderately with longer IESs and OESs. This suggests that for most short DNA excision, interactions between PiggyMac and the other proteins and scnRNAs/iesRNAs are less important than for longer DNA excision. For comparison, the silencing effects upon these and related genes affecting Paramecium genome development, and the expression/localization patterns of their encoded proteins are summarized in Table 1 and Table 2, respectively.
Even though the knockdowns of DCL2/3/5, NOWA1/2 and TFIIS4 seem to affect short IESs in a very similar manner, they affect the development-specific sRNAs in quite different ways. DCL2/3/5-KD leads to elimination of both scnRNAs and iesRNAs; NOWA1/2-KD inhibits RNA scanning (MAC genome-matching scnRNAs are not properly removed) and iesRNA production; TFIIS4-KD has little, if any, effect upon RNA scanning, but does eliminate iesRNA production12. Though limited to four analyzed IESs, it is thought that TFIIS4 may be involved in general IES transcription in the developing new MAC12, including that of an IES whose excision has no apparent requirement for scnRNAs, iesRNAs or TFIIS412 (51A4404; see Table S1). This is proposed to produce RNA to which the sRNAs bind, facilitating IES targeting12. Since TFIIS4-KD also leads to OES retention, it is likely that TFIIS4 is also involved in extensive transcription of these regions. It is also apparent that even when there is a good correlation in IES or OES retention scores, the normal expression and localization patterns of tagged proteins encoded by the genes tested in these knockdowns may be quite distinct, e.g. Nowa1/2 proteins are present in both old and new macronuclei, whereas TFIIS4 is predominantly present in new macronuclei (Table 1).
At present, the simplest explanation which reconciles the observed correlations in IES and OES retention scores between gene knockdowns, the effects of these knockdowns on sRNAs, and the expression and localization of the proteins encoded by these genes is that: (i) TFIIS4-KD removes the RNA transcripts which both scnRNAs and iesRNAs need to bind to their target IESs (or OESs, in the case of scnRNAs), hence IES retention following TFIIS4-KD correlates well with that following DCL2/3/5-KD (and TFIIS4-KD OES retention correlates modestly with DCL2/3-KD); (ii) NOWA1/2-KD prevents binding of Piwi-bound scnRNAs and iesRNAs to their targets via the nascent RNAs produced by TFIIS4, hence NOWA1/2-KD IES retention correlates well with both TFIIS4-KD and DCL2/3/5-KD IES retention (and NOWA1/2-KD OES retention correlates well with both TFIIS4-KD and DCL2/3-KD IES retention).
For the shortest IESs (those < 35 bp), we propose that the high IES retention score correlations between DCL2/3/5-KD, NOWA1/2-KD, TFIIS4-KD, PTCAF1-KD and EZL1-KD, may be a consequence of cooperation of scnRNAs, iesRNAs, RNA transcripts and chromatin state changes allowing these genomic regions to be recognized and targeted (Figure 3B). For longer IESs the correlations in IES retention score between the different possible pairs of DCL2/3-KD, DCL2/3/5-KD, NOWA1/2-KD, TFIIS4-KD are all strong, as are those between PTCAF1-KD and EZL1-KD, but those between the latter two knockdowns and the former ones are modest (Figure 3C). This reflects that long IESs have a greater requirement for scnRNAs, Nowa1/2 proteins and TFIIS4-dependent RNA transcripts, and very little requirement for iesRNAs, but that only some IES retention due to PTCAF1-KD and EZL1-KD also occurs when either scnRNAs, Nowa1/2 or RNA transcripts are depleted.
Though potential co-operation between chromatin modifying genes and scnRNA/iesRNA genes in IES excision is suggested by the modest correlations between their knockdown-induced IES retention scores, much of the excision requiring the Ezl1 and PtCAF1 chromatin-modifying proteins does not appear to need development-specific sRNAs, as there is generally much less IES retention after DCL2/3/5-KD (median 0.053) than after EZL1-KD or PTCAF1-KD (median 0.37 and 0.23, respectively) even though DCL2/3/5-KD effectively eliminates the IES-matching sRNAs (Figure 5C). DCL2/3 knockdown abolishes most H3K27me3 and H3K9me3 during new MAC development11, like EZL1-KD, PTCAF1-KD and NOWA1/2-KD10,11, whereas DCL5-KD does not influence these modifications11. Thus it has been suggested that scnRNAs alone may guide histone modifications that facilitate DNA elimination in Paramecium11.
The idea that Paramecium’s scnRNAs guide chromatin state changes in the developing new MAC should, however, be interpreted cautiously: though both PTCAF1-KD and NOWA1/2-KD affect H3K27me3 and H3K9me3 histone modifications in the new MAC, both PtCAF1 and Nowa1/2 also localize in the actively expressed, old MAC, and both PTCAF1-KD and NOWA1/2-KD10 affect H3K9me3 and H3K27me3 and scanning of scnRNAs proposed to occur in this nucleus, showing that PtCAF1 and Nowa1/2 (and potentially also the chromatin modifications that they influence) operate upstream of any possible scnRNA-directed chromatin modifications in the new MAC. It will thus be important to determine whether, like PTCAF1-KD and NOWA1/2-KD, EZL1-KD influences RNA scanning and the scnRNA population. Assuming fairly accurate quantification, more IESs are very strongly retained following DCL2/3/5-KD than EZL1-KD or PTCAF1-KD, e.g. at a threshold of > 0.7, 4316 IESs for DCL2/3/5-KD, compared to 1077 and 649 IESs for EZL1-KD or PTCAF1-KD), suggesting there may be some scnRNA/iesRNA-dependent IES excision which does not require the chromatin state changes induced by Ezl1- and PtCAF1. In future, it will thus be necessary to determine how the silencing of chromatin modifying genes, such as EZL1 and PTCAF1, and the removal of histone modifications, prevent excision of scnRNA/iesRNA-independent IESs, and, reciprocally, how depletion of scnRNAs and iesRNAs by DCL2/3/5 silencing prevents IES excision independently of Ezl1- or PtCAF1-dependent histone modifications.
Previously, it was proposed that the mechanisms involved in imprecise DNA elimination in Paramecium are distinct from those involved in IES excision6. For imprecise DNA elimination, it was suggested that the resultant boundaries are a mixture of AT-rich direct repeats6. Since these repeats are an arbitrary length and are in relatively AT rich genomic regions it is straightforward to find them purely by chance: e.g. 39 out 40 of the proposed direct repeats bounding the imprecisely eliminated DNA contained a TA dinucleotide, with all 7 of the two nucleotide repeats being TA6. We examined additional junctions of imprecisely eliminated regions (OESs) in DNA from control cells and consistently found that where direct repeats were present and unambiguous, they invariably contained a TA dinucleotide. We therefore favor the simple explanation that, like IESs, the imprecisely eliminated OESs are excised by PiggyMac transposases, with TA dinucleotides as the invariant bases necessary for this cleavage47. We previously noted that long IESs in Paramecium tend to be underrepresented in MIC genome regions that develop into coding sequences in the MAC genome, suggesting counterselection against such IESs49, either due to their lower excision efficiency or higher excision imprecision. Supporting the latter, we find that IESs with evidence of alternative excision in their vicinity are longer than those that do not (Figure S5E).
For the correlations between pairs of gene knockdowns, the effects on short IESs can be seen to differ from those on long IESs, and OES retention correlations more closely resemble those of long IESs than those of short IESs. We therefore propose there is a continuum of excision precision, which is highest for the shortest IESs and lowest for the longest IESs and OESs. In this case, the abbreviation “IES” may be a misnomer, since excision of longer DNA may lead to chromosome fragmentation rather than internal elimination. In a model in which PiggyMac operates as a dimer1 that recognizes and cuts at two sites, for short IESs it is most likely that both subunits recognize and cut where expected. As IES length increases, DNA elimination imprecision increases. When IESs/OESs become sufficiently long, the two PiggyMac proteins become increasingly unlikely to make contact, or the cut sites may be too far apart to be rejoined efficiently (the latter, proposed by 6), thus imprecise chromosome breakage results. Thus, consistent with our overall observations, it follows that the recognition and delimitation of the longest eliminated DNA sequences, including transposons, benefits most from additional factors, such as sRNAs or chromatin alterations.
In conclusion, we have shown that comparisons of DNA excision readout following gene silencing by RNAi now provide the means to identify and study associations among genome editing components in Paramecium on a genomic scale. Using this approach, we have proposed putative associations between pairs of Dicer-like and Piwi proteins involved in DNA elimination, which were subsequently confirmed by experimental pulldowns of these proteins and their bound sRNAs48. In principle, the application of similar methods would also be useful in identifying and analyzing the molecular components of pathways in forms of developmental DNA deletion found in diverse eukaryotes, including that occurring in parasitic nematodes50, sea lamprey51, and copepods52. In the case of the parasitic Ascaris nematodes, this includes DNA elimination which is considered unlikely to have a requirement for development-specific sRNAs such as those in ciliates53, but which could conceivably require specific chromatin modifications. As highlighted by the analysis of NOWA1/2 knockdown effects in this paper, these methods, together with complementary experiments, set the stage to begin generally disentangling interactions between candidate genome editing genes.
Datasets 1 and 2, IES and OES retention scores, respectively, are available from Zenodo: DOI, 10.5281/zenodo.82376654.
The latest source code of after_ParTIES is available from GitHub: https://github.com/gh-ecs/After_ParTIES.
The archived source code as at the time of publication is available from Zenodo: DOI, 10.5281/zenodo.83223655.
License: BSD 2-Clause License.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)