Keywords
Hepatic encephalopathy, acute liver failure, chronic liver injury, neuroinflammation, pathogenesis
Hepatic encephalopathy, acute liver failure, chronic liver injury, neuroinflammation, pathogenesis
Advanced liver disease is well known for its systemic consequences, notably its profound effects on brain function, in its most classic form, known as hepatic encephalopathy (HE). HE is currently defined as a brain dysfunction secondary to liver insufficiency and/or portosystemic shunting that manifests as a broad spectrum of neuropsychiatric abnormalities ranging from subclinical alterations to coma1. It remains a diagnosis of exclusion1. HE is frequent: overt HE is found in some 30–40% of patients with cirrhosis1. In 2009, HE resulted in 22,931 hospitalizations with an average cost of each stay ranging from $46,663–$63,1082. This highlights both the frequency of HE and the economic burden it currently places on the US.
The broad spectrum of HE has led to the development of multiple grading and classification systems to better categorize the severity of disease. It is currently a recommendation by the American Association for the Study of Liver Disease (AASLD) that HE should be classified according to the type of underlying disease, severity of manifestations, and precipitating factors1. Types A, B, and C of HE define the underlying disease. Type A (acute) is due to acute liver failure (ALF), type B (bypass) is due to portosystemic shunting without intrinsic liver disease, and type C (cirrhosis) is due to cirrhosis1. Each type has differing symptoms at presentation owing to the varying etiologies.
ALF, with the original term being fulminant hepatic failure, is defined as severe liver injury in the absence of pre-existing liver disease. The etiology of ALF is complex, ranging from viruses and drugs to genetic causes, with a vast majority remaining idiopathic3. In developing nations, hepatitis (A, B, and C) is the most common cause of ALF, while in the US, acetaminophen toxicity accounts for 39% of ALF cases3. Physical exam findings are often non-specific and generally reflect severe underlying liver dysfunction or complications thereof. ALF is currently a clinical diagnosis, made in the setting of acute liver injury in the context of abnormal liver tests (including dramatically elevated aminotransferases, and most importantly evidence of hepatocyte dysfunction demonstrated by a prolonged PT and increased INR3). Unique to type A is its association with increased intracranial pressure (ICP), thus carrying a risk for cerebral herniation1. It is believed that increased ICP results from cerebral edema secondary to hyperammonemia3. One current proposed mechanism is that in ALF the development of hyperammonemia outpaces the compensatory mechanisms seen in chronic liver disease3. Subsequently, ammonia increases intracellular osmolarity when it is metabolized to glutamine, resulting in cerebral edema causing increased ICP3.
Type B HE is secondary to portosystemic bypass or bypass with no intrinsic hepatocellular disease. Precipitating factors of HE in type B are factors that are known to increase ammonia, such as azotemia, infection, GI bleed, lactulose noncompliance, and constipation4. The most serious complication of transjugular intrahepatic portosystemic shunts (TIPS) is chronic recurrent HE that is refractory to standard treatment4. If severe enough, shunt revision may be warranted4.
Type C HE is associated with liver cirrhosis accompanied with either portal hypertension or portosystemic shunts1. The causes of cirrhosis are vast, ranging from viral, autoimmune, chronic biliary disease, and fatty liver diseases to rare storage diseases such as hemochromatosis and Wilson’s disease5. Signs and symptoms of cirrhosis are often present and include spider angioma, muscle wasting, jaundice, ascites, and gynecomastia and testicular atrophy in men. The most common form of HE in type C is minimal HE (MHE), which affects nearly 80% of cirrhotics6. MHE affects the patient’s daily life, interfering with executive function including their working memory and orientation6. Physical examination in MHE is often normal, and patients may present with subtle abnormalities that can be diagnosed by experts using specialized neurophysiologic tests, such as the critical flicker frequency test (CFF)6. Currently, there is no gold standard test, and a number of studies are currently investigating the efficacy and use of them7.
As stated above, HE is broken into three major types because of its multiple etiologies. HE presents with a broad spectrum of symptoms ranging from subclinical to comatose. This wide range of presentation is due to the multifaceted pathophysiology that underlies this complex disease. Highlighted below is the role of ammonia and inflammation in the development of HE.
Currently, ammonia is the best-characterized neurotoxin in the pathogenesis of HE and also appears to be important in the genesis of astrocyte swelling8. In healthy individuals, nitrogenous compounds, such as proteins, are metabolized by gut microflora and transported to the liver in the form of ammonia8. In the liver, ammonia is metabolized by the urea cycle with the majority of the subsequent urea excreted renally8. Advanced liver disease or portosystemic shunting leads to a buildup of ammonia in the blood8. Ammonia that builds up in the blood is then able to cross the blood–brain barrier, where it is metabolized by astrocytes into glutamine8. Hence, levels of glutamine start to accumulate and lead to astrocyte swelling. This swelling can trigger a downward spiral leading to increase in production of reactive oxygen and nitrogen species, which can downstream target gene transcription and translation9. Animal models of HE emphasize that astrocyte and brain swelling is also a key feature. Among these features are swelling of astrocytes, vasoconstriction of blood vessels, increase in ICP, cerebral edema, reduced cerebral perfusion, and cerebral atrophy9. There are conflicting studies regarding the correlation of levels of ammonia and the degree of encephalopathy; however, evidence suggests that a reduction in levels of ammonia leads to reduced brain swelling9.
In addition to the ammonia hypothesis, inflammation and cytokines are thought to be major components in the development of HE, particularly in the setting of ALF with or without sepsis10. Septic encephalopathy is a well-documented phenomenon that closely resembles HE10. Although sepsis and ALF have varied pathogenic mechanisms, they share the same cardinal features of encephalopathy, cardiovascular collapse, and coagulopathy10. Both sepsis and ALF result in an upregulation of inflammatory cytokines IL-1β, IL-6, and TNF-α10. Studies have shown that these inflammatory cytokines compromise the blood–brain barrier and disrupt the permeability of the endothelial cells10. It is not surprising that sepsis can precipitate and worsen HE10. Cytokines, infection, and inflammation play a significant role in the development of HE. Studies have implicated both ammonia and inflammation in the pathogenesis of HE10. Currently, it is proposed that these pathways have a synergistic effect on each other10.
Recent advances in the understanding of the pathogenesis of HE focus on the consequences of hyperammonemia and altered neurotransmission, and new studies have aimed to examine new potential targets such as bile acid signaling and their contribution to HE. Below is a summary of the latest findings (see also Figure 1).
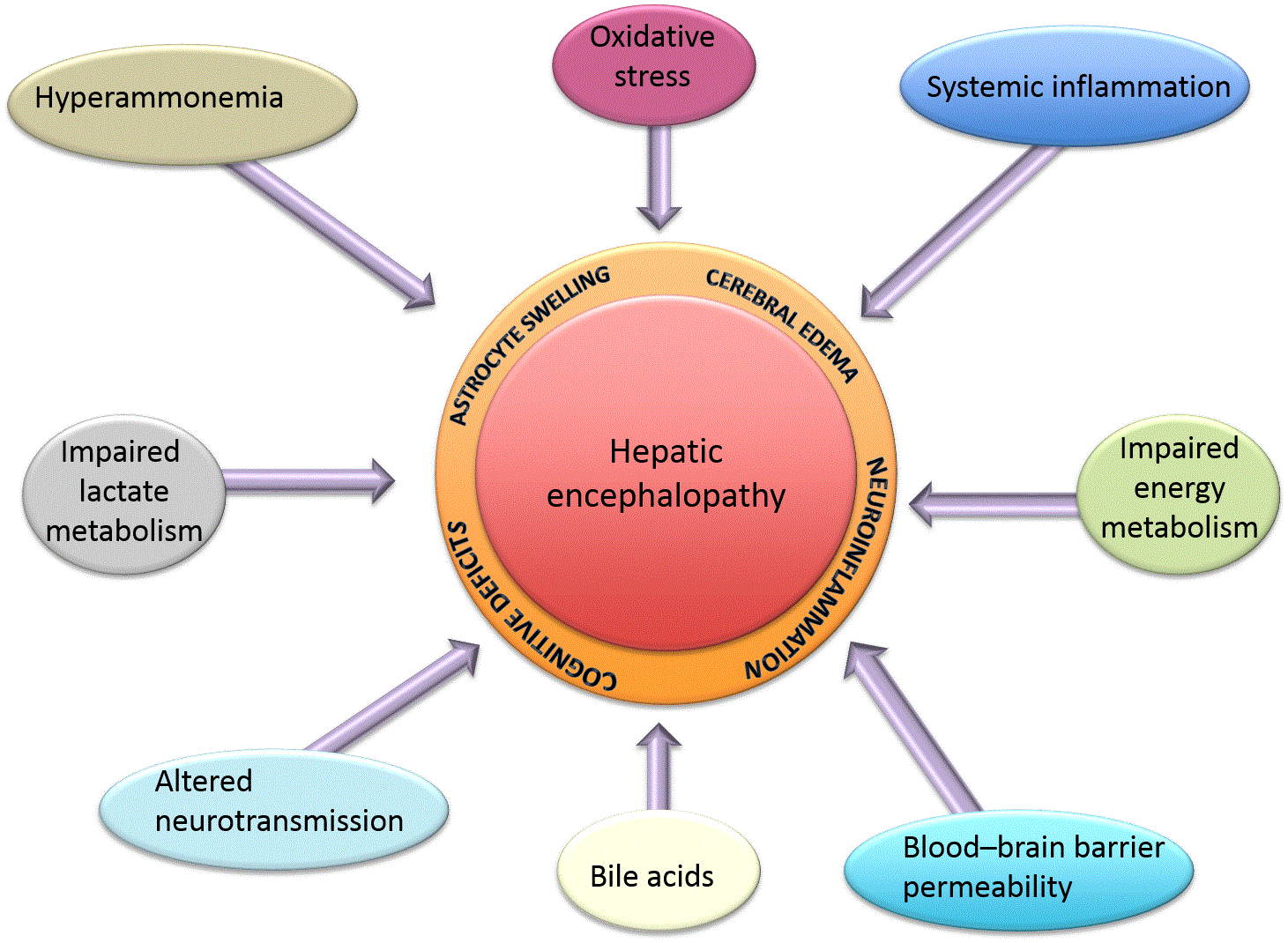
A major contributing factor to the development of HE is a buildup of ammonia. Liver-specific glutamine synthetase knockout (a key enzyme in ammonia metabolism) led to dramatically increased systemic hyperammonemia and subsequent cerebral oxidative stress and cognitive changes11, consistent with the concept that the liver is integral in the maintenance of ammonia homeostasis throughout the body.
In the brain under hyperammonemic conditions, astrocytes rapidly convert blood-derived ammonia into glutamine, increasing glutamine levels12. An understanding of the full array of consequences of hyperammonemia on astrocytic function in the context of HE is, as yet, unappreciated. Historically, it was thought that the effects of increased cerebral ammonia were increased oxidative stress, osmotic pressure, and subsequent astrocyte swelling. However, recent studies have demonstrated that ammonia also has effects on many signal transduction pathways13–15, gene expression16, and post-translational protein modifications17, which together lead to subsequent impairment in astrocyte function and manifest as abnormal proliferation18,19, neurotransmitter release16, and even cellular senescence18. For example, hyperammonemia was recently associated with a transient increase in astrocyte intracellular calcium release13–15; the precise mechanism of action may be through activation of NMDA receptor13, the transient receptor potential channel 114, or the Cav1.2 L-type calcium channel15, or, more likely, a combination of all of the above. Given that calcium-mediated effects are an early event of many different signal transduction pathways, the vast array of potential gene expression alterations due to hyperammonemia has not yet been defined.
A relatively novel mechanism by which ammonia may influence gene expression is via alterations in microRNA expression. MicroRNAs are small non-coding RNA sequences that play a role in gene silencing at the level of gene transcription as well as translation and can be regulated by oxidative stress. Exposure of astrocytes to ammonia dramatically altered microRNA profiles19, leading to an alteration in many target genes, including heme oxygenase 1. Preventing ammonia-induced microRNA expression changes increased the expression of heme oxygenase 1 and prevented the ammonia-induced inhibition of astrocyte proliferation and prevented the associated cellular senescence19.
Lastly, though not surprisingly, ammonia has been shown to alter the expression of genes involved in the regulation of astrocytic glutamate uptake and neurotransmitter release, such as the ephrin receptors and their ligands16 and thrombospondin 120. Specifically, the treatment of astrocytes in culture with ammonia increased ephrin receptor 4 via an as-yet-undefined pathway that includes glutamine synthetase, NADPH oxidase, and nitric oxide synthase activities16. Conversely, chronic ammonia treatment decreased thrombospondin 1 expression in astrocytes, which subsequently decreased the expression of synaptic proteins in neurons20.
Ammonia may also contribute to the cell-to-cell crosstalk that occurs in HE. Specifically, the treatment of brain endothelial cells with ammonia, a mixture of cytokines, or lipopolysaccharide appeared to release factors that contribute to astrocyte swelling via the activation of Toll-like receptor 421. The understanding of cell–cell interactions and communication and the role of ammonia in HE is still quite limited. In further support of the notion that cell–cell communication is involved in ammonia-induced events during HE, strategies to uncouple the gap junctions in astrocyte cultures significantly attenuated key features of ammonia-induced neurotoxicity, such as membrane integrity, oxidative stress, and pro-inflammatory cytokine release22.
Oxidative stress and the generation of reactive oxygen and nitrogen species in the brain has long been associated with HE. It is thought that impaired mitochondrial function and downregulation of the expression of key anti-oxidation enzymes contribute to an increase in oxidative damage to membrane lipids, protein, and DNA23.
Recent studies have emphasized the importance of oxidative damage in the pathogenesis of HE, with various experimental treatments aimed at reducing reactive oxygen/nitrogen species24–31 or restoring the activity of anti-oxidative enzymes such as catalase32, superoxide dismutase32, thioredoxin25, or glutathione peroxidase25. Of particular note was an elegant study by Bosoi et al.33, in which the authors utilized a rodent model of hyperammonemia that was also treated with an inhibitor of glutathione; they found that a synergistic relationship between systemic oxidative stress and hyperammonemia was required for the development of brain edema in HE33.
The pathogenesis of HE has long been associated with impairment in cerebral energy metabolism with alterations in glucose utilization, glycolysis, and mitochondrial dysfunction (reviewed in Rama Rao and Norenberg34). Of particular interest are the changes in cerebral lactate levels during HE. Specifically, increased blood and cerebral lactate levels have been demonstrated in patients with HE35, in pig and rodent models of ALF36,37, and in acute hyperammonemia38. However, the increase in cerebral lactate in models of type C HE is not as clear-cut. Increased lactate has been observed in the plasma of patients with cirrhosis compared to age- and sex-matched controls39. Interestingly, there was no significant difference in plasma lactate levels between cirrhotic patients who exhibit symptoms of overt HE versus patients without overt HE, although the presence of MHE in the latter group could not be ruled out39. A parallel change in lactate levels in the brain was not evident in a longitudinal study using 1H and 31P magnetic resonance spectroscopy in a rodent model of type C HE12. Conversely, de novo synthesis of lactate was increased in the brain, and treatment with a lactate synthesis inhibitor attenuated HE-associated brain edema in this model40. This disparity between these two opposing observations may lie in the methodology used to detect the differences. However, recently, it was demonstrated that the transport of lactate through connexin-containing hemichannels in astrocytes was impaired in the cerebral cortices of rats with type C HE owing, at least in part, to the actions of hyperammonemia41. Given that the astrocyte–neuron lactate shuttle hypothesis suggests that lactate production in astrocytes is able to fuel and regulate neuronal activity42, it was hypothesized that the impairment of lactate transport through hemichannels may be contributing to the pathogenesis of HE41.
Brain inflammation (i.e. “neuroinflammation”) is a key feature in common with all types of HE and appears to be predominantly modulated by microglia, the resident macrophage-like cell in the brain. Indirect clinical evidence for microglial activation has been demonstrated by an upregulation of the microglial marker ionized calcium-binding adaptor molecule 1 (Iba-1), which was found to be increased in post mortem cortical brain tissue from patients with liver cirrhosis and HE compared to cirrhotic patients without HE43. In addition, a comprehensive gene expression profile analysis showed that markers for both the pro-inflammatory M1 and the anti-inflammatory M2 microglial phenotypes were increased, suggesting that both groups can be found in patients with HE caused by cirrhosis44.
The activation of microglia is a delicate balance between the pro-inflammatory and anti-inflammatory signals, which in physiological conditions favors the dampening of microglia activation45. These signals may be derived from the microglia themselves or are as a result of cell-to-cell communication derived from neurons or astrocytes. Recently, the pro-inflammatory chemokine CCL2 was demonstrated to be increased in neurons in a mouse model of type A HE46 and a concomitant decrease in the anti-inflammatory chemokine fractalkine45, thereby dysregulating the balance between opposing pro- and anti-inflammatory signals acting on receptors on microglia resulting in microglia activation. Strategies to either block CCL2 receptors or increase fractalkine signaling inhibited the microglia activation and attenuated the cognitive dysfunction observed in this model of HE, although the precise mechanism by which the balance between these two opposing signals is dysregulated was not identified.
Interestingly, in the hyperammonemic rat model, microglia and astrocytes were activated with a concomitant increase in the expression of pro-inflammatory cytokines IL-1β and IL-647, suggesting that ammonia alone is capable of inducing neuroinflammation during HE, although it is unlikely that the modulation of neuroinflammation is solely the consequence of hyperammonemia during HE.
Evidence to suggest a causal link between neuroinflammation and cognitive and motor function impairment during HE is mounting. Strategies that specifically target and dampen the neuroinflammatory signals also offer attenuation of cognitive and motor deficits46,48–52, although care should be taken when interpreting data from experiments where the anti-inflammatory experimental agent is given systemically, as the mechanism of action may be via hepato-protection, thereby leading to a reduction in HE symptoms rather than as a direct modulatory effect on central neuroinflammation. While treatment strategies aimed at dampening systemic inflammation may be beneficial for both the underlying liver damage and the subsequent encephalopathy, from a basic science standpoint aimed at elucidating the pathogenic pathways associated with the development of HE, the distinction between the actions of an experimental compound on the brain versus its actions on the liver should be distinguished.
The cognitive and neuromuscular deficits observed during HE are ultimately the result of altered neurotransmission, regardless of the mechanism53. Interestingly, there are opposing effects on glutamatergic neurotransmission depending upon the type of HE with increased extracellular glutamate levels observed after ALF, and a dampened glutamatergic neurotransmission observed during chronic liver disease53. During ALF, the activation of NMDA receptors on astrocytes downregulates the expression of Kir4.1, an inward rectifying potassium channel known to regulate ion and water homeostasis54 and contribute to neuronal dysfunction in other neurodegenerative diseases55. The precise role for Kir4.1 in type A HE is not clear. Furthermore, strategies to block NMDA receptors in rats with ALF reduced the HE-associated changes in cerebral blood flow and brain lactate as well as increased the kidney clearance of ammonia, which collectively delayed or prevented the HE-associated mortality56. Conversely, during MHE due to chronic liver disease, memory impairment was associated with suppression of the glutamate–nitric oxide–cyclic guanosine monophosphate (GMP) pathway, which has been demonstrated in response to hyperammonemia57 or increased intracranial dopamine58, and strategies to restore this pathway also restore the learning ability of rats with HE57.
Both type A and type C HE are associated with increased GABA-ergic tone. This is due to a number of factors: increased GABA concentrations59, increased GABA receptor expression59, and increased neurosteroids such as allopregnanolone, known to regulate GABA-ergic neurotransmission60. Neurosteroids are steroids produced de novo in the brain61. Their production and function are currently under investigation. Recently, experimental compounds aimed at either inhibiting neurosteroid synthesis62 or inhibiting the enhanced activation of GABAA receptors by neurosteroids63 attenuated the deficits in motor co-ordination, spatial memory, and circadian rhythms observed during HE62,63. These data were supported by the use of an inhibitor of another neurotransmitter system thought to be dysregulated during at least MHE: the dopamine system64. However, how this system is altered during HE remains unclear. It has been hypothesized that an increase in dopamine release from cirrhotic liver into the bloodstream contributes to the pathogenic features of MHE by inhibiting the glutamate–nitric oxide–cyclic GMP pathway in the hippocampus, thereby altering learning and memory ability58,64–66. Conversely, dopamine and serotonin levels were decreased in another model of MHE67.
Elevated circulating and cranial levels of bile acids have been observed in patients with HE as early as 1977, although our understanding of the implications of this observation are only just coming to light. Specifically, total bile acid content in the cerebrospinal fluid as well as in brain tissue was elevated in patients and rodent models of type A HE68,69 as well as type C HE70–72; however, the precise role of bile acids in HE remains controversial. Increased serum bile acids have been implicated in the increased blood–brain barrier permeability observed in a rat model of chronic liver disease70, thereby allowing access of bile acids and other signaling molecules to the brain. In mice, azoxymethane (AOM) is used as a model for type A HE due to ALF69. Indeed, in the AOM mouse model of type A HE, increased total bile acid content was observed in the frontal cortex, and strategies to reduce circulating bile acids (e.g. cholestyramine feeding or the use of a genetically modified mouse with impaired bile acid synthesis) proved neuroprotective69.
Bile acids can exert their effects via many different receptors that result in an array of physiological responses. FXR is a nuclear bile acid receptor, and inhibition of FXR-mediated signaling in the frontal cortex conferred partial protection against the cognitive deficit that occurs during HE69. Furthermore, aberrant bile acid signaling in the brain can also affect neuroinflammation by increasing pro-inflammatory CCL2 expression in neurons via a sphingosine-1-phosphate receptor 2-dependent mechanism, which subsequently leads to the activation of microglia and increase in pro-inflammatory cytokine expression73. Conversely, TGR5 is a membrane-bound G-protein-coupled bile acid receptor, the expression of which has been shown to decrease in the brain in conditions of hyperammonemia74. Activation of this receptor with specific agonists reduced the microglia activation and pro-inflammatory cytokine production observed in a mouse model of type A HE75.
Many therapeutic strategies to treat HE, such as lactulose and metronidazole, abolish gut microbiota with the aim of reducing ammonia production while also influencing the conjugation of secondary bile acids. By altering the bile acid pool, they generate protective effects by altering bile acid signaling. In support of this notion, feeding mice a diet enriched in particular bile acids, which does not alter the total bile acid pool but alters the balance of individual bile acids, changed the susceptibility of mice to the development of HE69. Specifically, mice fed a diet enriched with deoxycholic acid or cholic acid had significantly quicker neurological decline compared to control-fed or ursodeoxycholic acid-fed mice69. These data raise the possibility that the elevation of cranial bile acids as well as a change in specific bile acids in the bile acid pool contribute to the neurological decline associated with HE; further clinical studies that manipulate bile acids are necessary to determine whether these effects will be recapitulated in patients with HE.
The role of blood–brain barrier permeability is controversial in HE. No robust evidence of its breakdown exists; however, it is highly likely that bile acids and pro-inflammatory cytokines may affect its permeability70,76–79. In the AOM model of type A HE, permeability of the blood–brain barrier has been demonstrated as a late event during the development of HE78,79. Conversely, no changes in permeability could be detected by another group unless the mice were co-treated with trace amounts of lipopolysaccharide80, suggesting a synergistic role for inflammation with other key HE-related features in the changes in blood–brain barrier permeability. Consistent with this notion was a study demonstrating that circulating transforming growth factor-β (TGFβ) may contribute to the increased permeability of the blood–brain barrier in ALF78.
Evidence to suggest that blood–brain barrier permeability is increased in type C HE also exists. As stated above, increased permeability in the bile duct ligation model of HE is apparent and can be attributed, at least in part, to increased serum bile acids70. Associated with this increased permeability was a decrease in expression of tight junction proteins76.
HE remains a diagnosis of exclusion with a broad spectrum of clinical manifestations. The multifaceted presentation poses many limitations to the diagnosis of HE. Guidelines by the AASLD emphasize the use of specialized psychometric tests in the diagnosis of HE81–83. A multitude of tests exist, with some of the most studied being the psychomotor hepatic encephalopathy score (PHES), CFF, Stroop effect test, and electroencephalogram (EEG)81,84.
PHES is a battery of pencil and paper tests aimed at diagnosing HE85. PHES was studied in Turkey and Romania in order to standardize a normal value in a healthy population and also assess its utility85,86. Age and educational status were shown to affect score85.
Apart from PHES, a newly developed electronic number connection test (eNCT) was investigated by Wuensh et al.87. This test flashes the numbers 1–25 on a screen and requires the participant to click them in order while being timed; those patients with HE were found to be much slower87. This test promises to be an easier and faster method than PHES87.
In the CFF, patients are shown a light flicker that progressively decreases in frequency88. They must identify the frequency at which the light flickers88. CFF can be used as a measure of cortical function and the diagnosis of HE88. Studies show that with a cut-off value of <39 Hz, CFF has a sensitivity of 39%, specificity of 82%, and diagnostic accuracy of 70.6% for MHE82,88. CFF is an important and simple tool for diagnosis and should be combined with the model for end stage liver disease (MELD) score for better diagnosis89. MELD is a scoring system used to grade the severity of liver disease90.
A prospective study of 117 consecutive patients with cirrhosis examined the administration of the CFF to detect HE91. The authors found that MHE was associated with a reduced 5-year survival rate in patients with cirrhosis91. CFF could be used in the future to improve the accuracy of prognosis in patients with cirrhosis91.
Another study focused on Stroop testing, which tests psychomotor speed, attention, and cognitive flexibility92 in the form of an app coined EncephalApp. The app’s reliability was tested compared to the gold standard tests and was validated by multicenter and multi-national analysis in the USA with over 800 subjects92. Another validation of the EncephalApp was done with 437 cirrhotics and 308 controls, and it was compared with PHES and the inhibitory control test (ICT)93. When compared against the controls, the app was found to be equivalent in predicting HE, thus proving to be a convenient and validated method for the diagnosis of HE93.
Currently, the analysis of EEG patterns in children is used for grading HE in ALF94. Nonetheless, EEG use in HE remains controversial. One study recorded EEGs in 69 healthy controls and 113 adults with cirrhosis95. New spectral thresholds were calculated and optimized the performance of EEG for the diagnosis of HE95. Though improvements were made in the use of EEG for the diagnosis of HE, validation is needed along with further research to understand the pathophysiological mechanism behind the EEG changes95–97.
Key changes in brain function have been studied with functional magnetic resonance imaging (MRI)98 in combination with arterial-spin labeling to enhance the detection of HE99. Connectivity status between 90 brain regions was assessed by measuring blood flow. HE showed impairment in the ganglia–thalamo–cortical circuits, with increased blood flow in the right putamen99.
Acknowledging the multiple etiologies of HE, researchers investigated if the cause of HE affected its presentation on imaging. Neurocognitive, biochemical, and brain MRI changes have been examined in patients with cirrhotic HE and extrahepatic portal vein obstruction (EHPVO) HE100. Serum studies showed no change in ammonia levels while cytokine levels were higher in cirrhotic HE100. Cirrhotic HE showed imaging changes that were absent in EHPVO HE100. Cirrhotic HE affected multiple brain sites ranging from the frontal lobe all the way to the brainstem100.
Spontaneous brain activity can also be measured by examining the amplitude of low-frequency fluctuation (ALFF) in the MRI101. It can be used as a biomarker for the detection of HE101. Upcoming research focuses on ALFF in combination with blood oxygenation level-dependent functional MRI, creating new opportunities for the diagnosis of HE102.
Cerebral blood flow is altered in HE. Regional cerebral blood flow was studied by comparing dogs with congenital portosystemic shunt with HE to controls using nuclear imaging103. It showed decreased perfusion of subcortical and temporal regions in dogs with HE103. This highlights the role that nuclear imaging, specifically single photon emission computed tomography, can play in helping diagnose HE103.
Metabolic profiling analyzes body fluids or tissues by measuring low-molecular-weight compounds using either 1H-NMR spectroscopy or mass spectrometry techniques104. Currently, it is used in hepatocellular carcinoma; however, its use in HE had not been previously studied104. Studies have shown that metabolic profiling can distinguish overt HE, but it cannot discriminate between differing grades of HE or according to the severity of underlying liver disease104.
The gut microbiome is implicated in the pathogenesis of HE105–108. Urease-producing bacteria predominantly lead to elevated levels of ammonia and serve as a target for many treatments109. A cross-sectional study set out to determine the species of gut microbiota predominant in HE and suggested that an absence of Blastocystis spp. led to dysbiosis.
Proton pump inhibitors (PPIs), a major treatment for gastroesophageal reflux, have been shown to be related to HE110–112. While the exact mechanism underlying this association is not known, it is currently thought to be due to alterations in the gut microbiome. PPIs have a known association with gut dysbiosis110–112. A case-control study of one million people in Taiwan followed for an average of nearly 14 years demonstrated that in patients with both cirrhosis and HE, 38% used a PPI112; all categories of PPIs, except rabeprazole, led to an increased risk of HE112. It is crucial to recognize the irreversible consequences that can occur with non-specific selection of treatment for reflux. Likewise, a meta-analysis showed that those with liver dysfunction who took a PPI had a higher risk of developing HE compared to those who did not take a PPI110.
Recognizing the importance of the gut microbiome hypotheses has led to study of the role of probiotics in HE113,114. A meta-analysis of randomized trials suggested that probiotics are more effective in decreasing hospitalization rates, improving MHE, and preventing progression to overt HE115. Similarly, a further meta-analysis by the Cochrane collaboration revealed that when compared to placebo, probiotics mildly improved recovery, decreased overt HE, improved quality of life, and mildly decreased plasma ammonia116–118. Of note, probiotics have no effect on mortality116–118. Because of the low quality of evidence, no direct conclusions can be made, as more research is needed116–118. A double-blind randomized placebo-controlled clinical trial studied the effectiveness of VSL#3119 in Chandigarh, India. This study demonstrated reductions in hospitalizations and severity of breakthrough episodes of HE as well as improvement in PHES and liver function in patients receiving VSL#3119.
Diet changes have been studied to determine their effects on cognitive function in patients with MHE with cirrhosis120. One study in 2016 compared patients prescribed a diet with a caloric restriction of 30–35 kcal/kg and a protein intake of 1.0–1.5 g of vegetable protein/kg to a control group for six months120. PHES was used to diagnose HE. They found that a higher proportion of patients on the prescribed diet had reversal of HE (71.1% versus 22.9%, p=0.001) along with a decreased incidence of overt HE in the treatment group (10% versus 21.7%, p=0.4)120.
Alteration in the gut–brain axis in HE has led to investigations in determining the effectiveness of gut-selective antibiotics. Modulating the gut microbiota with the use of antibiotics such as rifaximin may improve cognitive performance in cirrhotics121–126. This hypothesis was tested in a study at the Hunter Holmes McGuire VA Medical Center127. Patients with confirmed HE were prescribed rifaximin 550 mg oral twice a day over a period of eight weeks127. Compared to the placebo group, the treatment group had cognitive improvement, decreased endotoxemia, and a modest decrease in bacteria of the small bowel127.
A retrospective, observational, cross-sectional study examined the efficacy of different rifaximin dosing (400 mg three times daily versus 550 mg twice daily)128. There was no difference in readmission rates between the two treatment groups128. The long-term cost effectiveness of lactulose monotherapy versus lactulose plus rifaximin was studied in France129. Despite having no effect on hospital length of stay, combination therapy led to lower readmission rates at 180 days compared to lactulose alone130. The authors concluded that rifaximin was a cost-effective and affordable treatment for patients suffering from at least two prior events of HE129.
As stated above, numerous studies have shown that ammonia is key in the pathogenesis of HE. Currently, the gold-standard treatment for the reduction of ammonia during HE is lactulose131, although a number of other experimental drugs aimed at lowering ammonia have been trialed including ornithine phenylacetate (OP)9,132–134 and L-ornithine L-aspartate (LOLA)135–137. A major breakthrough came with an investigation of bacterial-DNA translocation effects on hyperammonemia and neurocognitive scores in patients with HE after the use of lactulose131. The authors showed that neurocognitive scores improved with lactulose use and also led to reduced bacterial-DNA translocation131.
Evidence is mounting that the use of polyethylene glycol (PEG), with or without lactulose, is effective. A randomized controlled trial in 40 patients compared the use of lactulose versus combined lactulose and PEG in cirrhotic patients admitted with HE138. There was a statistically significant decrease in length of hospital stay (in women)138. Surprisingly, there was no effect on ammonia levels138. Likewise, the HELP randomized trial found that, compared to lactulose, PEG led to more rapid HE resolution in the hospital and concluded that it was superior to standard lactulose therapy139.
Studies are investigating albumin transfusion as a possible treatment for HE140. A prospective randomized controlled trial investigated lactulose alone versus lactulose plus albumin140. Combination treatment of lactulose plus albumin led to a decrease in levels of ammonia, IL-6, IL-18, and TNF-α and other endotoxins140.
Studies are also investigating branched chain amino acid (BCAA) supplementation in advanced liver disease. The ratio of normal BCAAs to aromatic amino acids becomes reduced in HE141. Randomized controlled studies show that BCAA supplementation does not prevent the development of HE but may slow down the progression of hepatic failure141,142. Despite BCAAs having no effect on mortality, 16 randomized controlled clinical trials have shown that BCAAs decrease the signs and symptoms of HE143. Conflicting studies exist, with some stating BCAAs have no significant effect on HE144.
Recent studies have examined the use of corticosteroid administration in patients with acute liver injury and ALF145. A prospective observational study of 469 patients from 2004 to 2015 showed that high-dose therapy decreased the risk of HE, but the time to initiate therapy has yet to be established145.
As might be predicted, liver transplantation has been shown to improve the brain activity and cognitive function of patients with cirrhosis with or without HE146. Zhang et al. studied the effect of liver transplant on resting-state brain activity by quantizing the ALFF before and one month after transplant. Cognitive function improved in both groups with and without encephalopathy, showing transplant as a viable treatment option for HE146.
In a case report, a 73-year-old male presented with new-onset HE and was found to have a congenital portosystemic shunt with a normal liver histology. Correction of the shunt led to full reversal of his symptoms147. This highlights a surgically correctable cause of acute HE without underlying liver disease.
An increase in shunt blood bypassing the liver owing to portal hypertension can result in HE. Surgical interventions to close these shunts have been studied as a possible treatment for HE148. Percutaneous transhepatic obliteration (PTO) and percutaneous transhepatic sclerotherapy (PTS) are two methods used to close shunts in the setting of emergent variceal hemorrhage when other methods have failed148. PTO is performed by placing metallic coils in the afferent veins to reduce blood flow, while PTS is performed by injecting a sclerosing agent148. Both methods utilize an invasive transcutaneous intrahepatic puncture148. A study of 37 patients with variceal hemorrhage and intractable HE underwent PTO/PTS148. Blood ammonia levels greatly improved, dropping from 135 mg/dL to 65 mg/dL at six months, and there was also an improvement in HE148.
TIPS is a procedure that places a stent between the hepatic vein and portal vein to alleviate portal hypertension149. However, allowing blood to bypass the liver can worsen HE149,150. The size of the stent in TIPS has been associated with HE151,152. One study showed that the use of a larger diameter stent improved ascites without a significant change in HE151,152. Additionally, the consumption of a low-protein diet after TIPS can reduce the incidence of HE153. Interestingly, antegrade embolization of spontaneous splenorenal shunt has also been employed to decrease the incidence of post-TIPS HE and has decreased the rate of variceal bleeding154.
Balloon-occluded retrograde transvenous obliteration (BRTO) is an alternative or adjuvant to TIPS in the management of gastric varices155. It works by abolishing gastrorenal or splenorenal shunts155. A retrospective record-based study of patients who underwent BRTO for gastric variceal bleeding or refractory HE showed great improvement in grade of encephalopathy after BRTO155.
This review summarizes the advances made in basic and clinical research over the last three years that have furthered our understanding of the pathophysiology of HE and possible treatments. Recent developments in basic research have focused on the role of hyperammonemia, oxidative stress, neuroinflammation, neurotransmitter function, bile acids, and the blood–brain barrier. More research is needed in these areas to fully establish the mechanism behind these various components and the role they play in the pathophysiology of HE. The diagnosis of HE is being investigated and is expected to become more precise by optimizing and validating current scoring systems. It appears that at least some of these will be amenable to modern app technology. Research in non-invasive imaging is also active and promises to aid in the diagnosis of HE. Strides have been made in the treatment of HE by specifically targeting the gut microbiome and hyperammonemia.
AASLD, American Association for the Study of Liver Disease; ALF, acute liver failure; ALFF, amplitude of low-frequency fluctuation; AOM, azoxymethane; BCAA, branched chain amino acid; BRTO, balloon-occluded retrograde transvenous obliteration; CFF, critical flicker frequency test; EEG, electroencephalogram; EHPVO, extrahepatic portal vein obstruction; GMP, guanosine monophosphate; HE, hepatic encephalopathy; ICP, intracranial pressure; MELD, model for end stage liver disease; MHE, minimal hepatic encephalopathy; MRI, magnetic resonance imaging; PEG, polyethylene glycol; PHES, psychomotor hepatic encephalopathy score; PPI, proton pump inhibitor; PTO, percutaneous transhepatic obliteration; PTS, percutaneous transhepatic sclerotherapy; TIPS, transjugular intrahepatic portosystemic shunts.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)