1Department of Molecular Physiology & Biophysics, Vanderbilt University School of Medicine, Nashville, TN, USA 2Vanderbilt Mouse Metabolic Phenotyping Center, Vanderbilt University School of Medicine, Nashville, TN, USA
OPEN PEER REVIEW
REVIEWER STATUS
Abstract
Obesity and insulin resistance often emerge from positive energy balance and generally are linked to low-grade inflammation. This low-grade inflammation has been called “meta-inflammation” because it is a consequence of the metabolic dysregulation that can accompany overnutrition. One means by which meta-inflammation is linked to insulin resistance is extracellular matrix expansion secondary to meta-inflammation, which we define here as “meta-fibrosis”. The significance of meta-fibrosis is that it reflects a situation in which the extracellular matrix functions as a multi-level integrator of local (for example, mitochondrial reactive oxygen species production) and systemic (for example, inflammation) inputs that couple to cellular processes creating insulin resistance. While adipose tissue extracellular matrix remodeling has received considerable attention, it is becoming increasingly apparent that liver and skeletal muscle extracellular matrix remodeling also contributes to insulin resistance. In this review, we address recent advances in our understanding of energy balance, mitochondrial energetics, meta-inflammation, and meta-fibrosis in the development of insulin resistance.
Corresponding author:
David H. Wasserman
Competing interests:
The authors declare that they have no competing interests.
Grant information:
The author(s) declared that this work was funded by National Institutes of Health (grants DK054902, DK050277, DK059637), NIDDK Mouse Metabolic Phenotyping Centers MICROMouse Program (grant 15GRU2558) and American Heart Association (grant 16POST29910001).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Advances in industrial and agricultural technology combined with lower rates of energy expenditure through physical activity have had the unintended consequence of creating a dramatic rise in the prevalence of obesity, insulin resistance (IR), hypertension, and dyslipidemia. These comorbidities are principal components of the metabolic syndrome as well as risk factors for type 2 diabetes mellitus and cardiovascular disease. The public health impact of these altered metabolic states is clear when considering that, in 2012, approximately 33% of United States citizens (over 100 million people) were projected to have at least one component of the metabolic syndrome1.
Positive energy balance at the whole-body level and altered oxidative metabolism at the cellular level are central to the development of IR. However, the conduit linking nutrient status and cellular energetics to pathophysiological states like IR is incompletely defined. In this commentary, we provide a framework for how mitochondrial energetics along with metabolically driven inflammation (meta-inflammation) and extracellular matrix (ECM) remodeling leading to fibrosis (meta-fibrosis) link overnutrition to IR (Figure 1). As several recent discoveries suggest, there is a great deal to be learned regarding the etiology of IR by studying organ-level physiological events in the context of the extracellular milieu. The focus here will be on metabolism, molecular organization, and cell signaling in the pathogenesis of IR. The important roles of gene transcription and epigenetics in the development of IR are beyond the scope of this commentary. Readers are directed to recent reviews on these topics2,3.
Figure 1. Positive energy balance promotes insulin resistance via metabolism-driven inflammation and fibrosis.
Energy balance is defined as the difference between absorbed dietary macronutrients (Supply) and energy expenditure (Demand). Energy supply is determined by the quantity and composition of macronutrients consumed, whereas energy demand is determined by exercise, non-exercise activity thermogenesis, and resting metabolic rate. A net positive energy balance (Supply > Demand) leads to obesity and a cascade of events that includes mitochondrial carbon stress (that is, an oversupply of macronutrients to mitochondria). This metabolic stress on mitochondria can promote meta-inflammation and meta-fibrosis that ultimately contribute to cellular and systemic insulin resistance.
Energy balance and the metabolic syndrome
Energy balance is defined as the gastrointestinal absorption of dietary macronutrients minus whole-body energy expenditure. Human evolution has selected for traits that facilitate the efficient mobilization, metabolism, and storage of macronutrients. The biological significance of these adaptations lies in the need to store nutrients during times of nutrient excess and the ability to mobilize fuel in situations of nutrient deficiency. Nutrient storage is important for acute bouts of elevated energy expenditure or prolonged periods during which food is not readily available. Indeed, mechanisms for storing excess glucose (glycogen), lipids (triglyceride), and amino acids (protein) obtained from the diet are exquisitely sensitive. While these adaptations have been critical for survival and species propagation, people living in industrialized societies now have easy access to high-calorie foods and do not need to expend considerable energy to obtain their food. This has led to a sustained positive energy balance. Since this is a situation rarely encountered during the course of human evolution, the body is poorly equipped to adapt to dietary excess. As such, the chronic energy surplus incurred by overnutrition and sedentary behavior has become a persistent metabolic burden that leads to adipose tissue expansion and obesity in many individuals4. Obesity, in turn, is central to the development of IR.
The evolutionarily conserved mechanisms that make survival possible during periods of famine also make humans refractory to weight loss. Resistance to weight loss and weight maintenance is recognized as a primary barrier to improving metabolic health5. This is most clearly demonstrated when considering the effects of caloric restriction and physical activity on energy balance and body weight. In both obese and non-obese humans6–8, prolonged caloric restriction results in significant weight loss, but it is accompanied by reductions in resting metabolic rate (RMR) beyond that which can be accounted for by weight loss alone. Since RMR is a primary contributor to the daily energy budget9, this represents a significant barrier to long-term weight loss. It is notable that exercise alone is only marginally effective as a therapy for weight loss10–12. This is likely due to both metabolic and behavioral obstacles. Exercise training fails to increase RMR in obese individuals with diabetes13, and this is potentially due to increased metabolic efficiency14. Exercise training has had mixed results in eliciting weight loss in both rodents15 and humans10 and is explained in part by increased food intake. In addition to RMR, “non-exercise activity thermogenesis” (NEAT) is a major contributor to energy expenditure in mice and humans16. Mice given access to a running wheel increase their physical activity and energy expenditure over a four-week period, but the metabolic cost of activity progressively decreases concurrently with decreased NEAT17. This is significant because fat gain with overnutrition in humans is positively correlated with an increase in NEAT18. Whether the bidirectional modulation of NEAT based on whole-body energy balance is modifiable therapeutically remains to be seen but may be a viable strategy for combating obesity. Complicating therapeutic strategies further is a growing body of literature demonstrating that the metabolic adaptations that occur with weight loss predispose an individual to accelerated weight regain and increased adiposity upon cessation of a supervised diet or exercise regimen or both19. Notably, recent work suggests that glucocorticoid antagonism mitigates the weight regain and IR that occur following cessation of voluntary exercise in rats20. A better understanding of how humans resist weight loss, even in the setting of obesity, is critically important in that it may reveal novel therapeutic strategies for treating obesity and IR.
Mitochondrial energetics and the pathogenesis of insulin resistance
As the demand-driven terminus of oxidative metabolism, mitochondria are intricately involved in the maintenance of energy balance, and several recent reviews have highlighted the importance of mitochondrial energetics to the etiology of IR21–23. At the level of the mitochondrion, energy balance is established by a dynamic rate of carbon flux through the tricarboxylic acid (TCA) cycle that supports ATP production via oxidative phosphorylation. In the setting of overnutrition, there is a supply/demand mismatch that results in excess anaplerotic flux of carbon from fatty acids entering the TCA cycle relative to the ATP demand leading to IR24. Excessive anaplerotic flux creates a mitochondrial “carbon stress” that has been well documented in both skeletal muscle (SkM) and liver. This carbon stress promotes IR through incompletely defined mechanisms that likely involve post-translational protein modifications that alter insulin signaling or protein trafficking (that is, GLUT4 translocation). The teleological explanation for limiting SkM glucose uptake in the face of excess dietary lipids may be that SkM is unable to efficiently convert excess intracellular glucose to an inert metabolite (that is, fatty acids).
In the liver, greater fatty acid availability accelerates anaplerotic flux contributing to IR that correlates with the severity of non-alcoholic fatty liver disease (NAFLD) in humans25. This appears to be linked, at least in part, to incomplete β-oxidation in the setting of overnutrition26. This hypothesis is supported by findings that acyl-carnitine, the carbon chain intermediate of β-oxidation, is increased in human plasma27 as well as rodent SkM26. Free carnitine in SkM is also reduced in the setting of obesity or high-fat feeding or both28, suggesting a reduced capacity to handle excess fatty acids. Collectively, excess dietary fatty acids entering metabolically active tissues overload the mitochondria, leading to IR. A teleological explanation for why mitochondria induce IR may be to mitigate oxidative damage induced by overnutrition29.
Mitochondria can also engage in cataplerosis, which is removal of carbons from the TCA cycle. In SkM, one proposed role for cataplerosis is as a buffering system to avoid mitochondrial carbon excess that can lead to increased reactive oxygen species (ROS) production during overnutrition24. SkM cataplerosis occurs in large part via carnitine acetyltransferase (CrAT), an enzyme that is responsible for exporting acetyl and acyl groups bound to carnitine from the mitochondrial matrix into the cytosol. Mice with SkM-specific deletion of CrAT have impaired glucose tolerance and increased oxidative stress30, illustrating a need for mitochondrial carbon efflux (that is, cataplerosis) to preserve SkM metabolic homeostasis in the setting of overnutrition. In the liver, cataplerosis is essential for the production of both glucose (gluconeogenesis) and ketones (ketogenesis). Predominantly expressed in gluconeogenic organs (liver and kidney), phosphoenolpyruvate carboxykinase (PEPCK) converts oxaloacetate to pyruvate and is a key enzyme for gluconeogenesis. Loss of PEPCK in mice reduces hyperglycemia in leptin receptor–deficient (db/db) diabetic mice31. Similarly, ketogenesis exerts partial protection against high-fat diet (60% calories from fat)–induced hyperglycemia and fatty liver, primary complications linked to obesity and overnutrition32. Notably, however, mice fed a ketogenic diet (more than 90% calories from fat) are lean and hypoinsulinemic but also display fatty liver33,34. This may be due to the impaired liver mitochondrial respiratory capacity observed in mice fed a short-term (14 days) ketogenic diet35. Strategies to increase cataplerosis in a tissue- and product-specific fashion could yield valuable strategies for preserving glucose homeostasis and insulin sensitivity but should be considered in the context of also preventing the development of fatty liver.
How does mitochondrial carbon excess promote IR? Carbon turnover that exceeds metabolic demand leads to accumulation of reducing equivalents (NADH and FADH2) that exert greater “reducing pressure” (that is, more electrons) on the electron transport system36. This buildup of reducing equivalents in the matrix and electrons within the electron transport system promotes the formation of ROS that modulate a wide variety of normal and pathophysiological cellular processes37. For example, acute or chronic high-fat feeding increases mitochondrial ROS production that has been shown in some29,38–40, but not all41, reports to be causal for the development of IR. Notably, fatty acids can also “uncouple” oxidative phosphorylation42, raising the possibility that mitochondrial oxidative efficiency may be an additional mechanism to manage carbon excess in obesity. Targeting this mechanism may be feasible in light of recent work demonstrating that mitochondrial oxidative efficiency is a dynamic process that is acutely sensitive to energetic demand43. Historically, the use of mitochondrial uncouplers as therapeutic agents has been met with skepticism following a string of deaths linked to the protonophore 2-dinitrophenol in the 1930s. However, recent efforts have provided new lead compounds that may be promising in the treatment of obesity44–46. While mitochondria-targeted therapies are being studied intensively and hold great promise, an alternative approach may be to address downstream effectors of mitochondrial oxidants. The downstream processes affected by mitochondrial oxidants are incompletely defined but include inflammation and expansion of the ECM. The remainder of this article will be spent discussing these processes in the context of their individual, and collective, contributions to the etiology of IR.
Inflammation and extracellular matrix expansion in the etiology of insulin resistance
Low-grade metabolically driven “meta-inflammation”47 contributes to IR in obesity48. There are numerous intersecting mechanisms linking inflammation and ROS49, including a critical role for the innate immune system that is coupled to macrophage infiltration50,51. Macrophages recruited with chronic overnutrition are pro-inflammatory (M1; CD11b+) and secrete tumor necrosis factor alpha (TNFα) that has been shown to contribute to IR in adipose, SkM, and liver52–54. M1 macrophages also play a critical role in wound healing. It has been observed that the meta-inflammatory response to obesity that includes M1 macrophage infiltration is responsible for the accumulation of ECM proteins in insulin-sensitive tissues55. The evidence linking these processes in adipose, SkM, and liver is outlined below.
Adipose tissue function is reliant upon, and in certain situations compromised by, the ECM surrounding adipocytes56. Healthy adipose tissue expansion involves a balance between enzymatic degradation and subsequent synthesis of ECM proteins57. Pathogenic obesity in humans is characterized by adipose tissue fibrosis due to excessive ECM deposition and reduced ECM degradation that is associated with IR58–61. Paradoxically, recent work by Muir et al.62 showed that diabetics have reduced adipose tissue fibrosis and greater adipose tissue hypertrophy. Genetically obese (ob/ob) mice have increased expression of genes encoding collagens63 that is exacerbated by high-fat feeding64. Genetic loss of the adipose tissue–abundant collagen VI in mice mitigates adipocyte inflammation, diet-induced obesity (DIO), and glucose intolerance while permitting greater adipocyte hypertrophy63. Beyond collagen, various other ECM components—including osteopontin65,66, hyaluronan67, thrombospondins68,69, and microfibril-associated glycoprotein 1 (MAGP1)70—accumulate in adipose tissue with obesity and contribute to IR. Adipose tissue ECM expansion is attenuated by the anti-diabetic drug metformin71, a drug that is also known to reduce mitochondrial ROS production39. Whether metformin improves metabolic health by mitigating mitochondrial ROS production or ECM accumulation or both remains to be addressed directly.
Obesity induces SkM ECM expansion55,72,73 that would be expected to increase the resistance to glucose delivery, an essential controller of glucose uptake74. Even short-term (28 days) high-fat feeding75 is sufficient to induce SkM ECM expansion. This appears to be reversible as SkM collagen accumulation is ameliorated in obese mice following exercise training73 and preventable in mice with genetic enhancement of SkM mitochondrial ROS scavenging55. A genetic knockout of matrix metalloprotease-9 (MMP-9), a key ECM-degrading enzyme, in obese mice causes increased collagen and a further deterioration of SkM insulin action76. Treatment with pegylated hyaluronidase causes degradation of hyaluronan and rescues IR in obese mice77. These studies demonstrate a direct link between ECM accumulation and insulin action in SkM.
In the setting of obesity, circulating lipids are incompletely sequestered in adipose tissue and consequently accumulate in SkM and liver and lead to IR. NAFLD is a primary risk factor for the development of IR and diabetes via liver fibrosis78,79. Mice fed a high-fat high-fructose diet exhibit liver fibrosis that accompanies lipid accumulation and IR80,81. The extent and scope to which overnutrition alters liver ECM are not completely known, highlighting a need for future studies.
ECM accumulation is recognized as a structural barrier between cells and the vascular space that restricts molecular transport82. More recently, a body of evidence has emerged indicating that cellular changes that accompany ECM accumulation are receptor-mediated. As such, the ECM is a biomolecular “motherboard” that determines the physical and metabolic properties of the tissue and the cells that they envelope. A greater understanding of how the ECM changes in obesity and the contribution of individual ECM proteins will be necessary in defining extracellular processes impacting metabolic health.
Extracellular matrix expansion and integrins in the setting of obesity
Integrins are a class of receptors that bind ECM proteins and have numerous overlapping functions, including cell adhesion, mechanotransduction, and differentiation83,84. ECM receptors are involved in a myriad of receptor signaling events through physical and functional interactions with growth factor receptors, including the insulin receptor85. In this way, ECM receptors orchestrate dynamic and specific signaling responses to diverse physiological and pathophysiological conditions. Integrins functionally link ECM changes to a multitude of conditions, including IR86 (summarized in Figure 2).
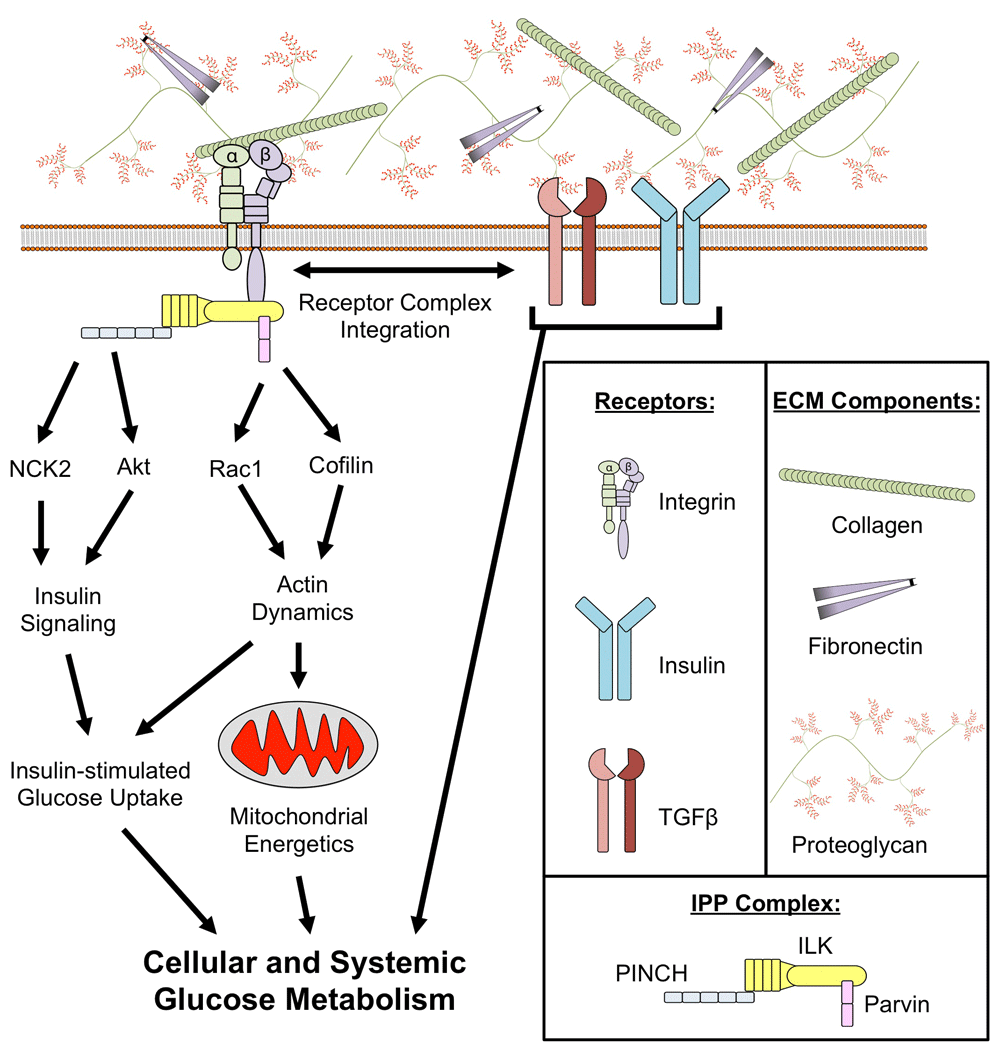
Figure 2. Putative mechanisms for the role of integrins in the development of insulin resistance.
Extracellular matrix (ECM) proteins are ligands for integrins, a family of cell surface receptors. Integrins are linked to the regulation of glucose metabolism through numerous mechanisms. Integrins can co-localize with transforming growth factor beta (TGFβ) and insulin receptors that are key regulators of glucose uptake into tissues. Integrins are also involved in intracellular signaling through the integrin-linked kinase (ILK)/PINCH/Parvin (IPP) complex. PINCH is characterized as a modulator of kinase signaling pathways as it regulates Nck2 and Akt, requisite proteins for insulin signaling. Parvin is involved in the regulation of cytoskeletal dynamics that permit remodeling and translocation of mitochondria and various intracellular proteins (that is, glucose transporters). The integration of integrins with regulatory nodes for glucose metabolism highlights the potential significance of ECM-integrin signaling in the etiology of insulin resistance.
Integrins are heterodimers consisting of α and β subunits with varying ligand specificities and expression in different tissues. Differentiated insulin-sensitive cells from SkM, adipose tissue, and liver express a variety of α subunit isoforms but express only a single β integrin isoform (β1)87–89. Whole-body loss of the integrin α1 subunit, a pro-fibrotic integrin receptor subunit that exclusively binds to β1, fails to protect against diet-induced SkM IR in mice; however, loss of the anti-fibrotic α2 isoform that also binds to β1 is protective55. It is interesting to note that combined SkM and myocardial loss of the integrin β1 subunit results in IR in lean mice90. Integrin-linked kinase (ILK) is a protein that physically associates with the intracellular tail of the β integrin subunit90. In contrast to the IR caused by knockout of the integrin β1 subunit in both SkM and myocardium of lean mice90, SkM-specific loss of ILK (mILK-KO mice) results in improved SkM insulin action in DIO mice91. Liver-specific deletion of ILK also protects against IR in DIO mice92. Whether adipocyte ILK deletion has effects on nutrient metabolism remains to be determined.
Despite its name, ILK lacks a functional kinase domain but rather functions as a scaffold for at least 26 high-fidelity binding partners93. Most notable among these binding proteins are PINCH and parvin, which, together with ILK, form an ILK/PINCH/Parvin (IPP) complex. PINCH consists of two isoforms (PINCH1 and PINCH2) that have both distinct and overlapping cellular functions94. In the context of glucose homeostasis, PINCH can bind to Nck2, which in turn interacts with insulin receptor substrate-1 (IRS-1)95, a requisite for insulin signaling. Nck2 is highly expressed in epididymal adipose tissue and its genetic deletion in mice causes IR and increased lipolysis96. PINCH has also been implicated in the phosphorylation of Akt via interactions with ILK97. Three ubiquitously expressed isoforms of parvin exist (α, β, and γ). α- and β-parvin both can bind directly to f-actin and in this way regulate cytoskeletal dynamics98. Parvin-mediated regulation of actin cytoskeletal dynamics is thought to occur, at least in part, via interactions between parvin and the Rho GTPase Rac199,100 and actin depolymerizing factor protein cofilin101. Rac1 is required for insulin-stimulated glucose uptake and is impaired during IR102, representing a potential link between integrins and insulin action. A potential role for γ-parvin in the context of insulin action has not been elucidated. Rac1 and cofilin are also involved in the regulation of numerous mitochondrial processes, including fission103, apoptosis104, and translocation105, demonstrating a link between integrins and the regulation of oxidative metabolism. Whether integrins and the IPP complex directly regulate Rac1 or cofilin in obesity is not yet known, nor is it known what role the IPP complex may play in obesity through its other binding partners. A recent report shows that focal adhesion kinase (FAK), an alternative downstream target of integrin activation, can modulate insulin sensitivity through regulation of adipocyte survival106. In light of the complexities of the ECM, integrins, and intracellular signaling pathways, much remains to be learned about ECM-integrin interactions in IR.
Summary and future directions
The etiology of IR involves both cell-intrinsic regulation of nutrient metabolism and integrated systems pathophysiology. The established paradigm of meta-inflammation coupled with the emerging concept of meta-fibrosis illustrates the complex nature of IR; however, several major questions remain to be addressed. For example, the composition and organization of the ECM must be elucidated so that the contribution of individual proteins or complexes or both can be mechanistically understood. Additionally, the role of downstream intracellular substrates of integrin signaling must be defined in the context of IR and cellular metabolism. The complex nature and broad importance of ECM/integrin function will be better understood through interdisciplinary studies that draw expertise from numerous fields (such as mechanobiology, biophysics, endocrinology, and molecular metabolism). It is anticipated that future studies will provide a more complete understanding of how the ECM functions as a biophysical regulator of whole-body function and support the development of novel therapeutics aimed at treating IR by mitigating meta-fibrosis.
Competing interests
The authors declare that they have no competing interests.
Grant information
The author(s) declared that this work was funded by National Institutes of Health (grants DK054902, DK050277, DK059637), NIDDK Mouse Metabolic Phenotyping Centers MICROMouse Program (grant 15GRU2558) and American Heart Association (grant 16POST29910001).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
3.
Gross B, Pawlak M, Lefebvre P, et al.:
PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD.
Nat Rev Endocrinol.
2017; 13(1): 36–49. PubMed Abstract
| Publisher Full Text
4.
Endorsed by The Obesity Society, Young DR, Hivert MF, et al.:
Sedentary Behavior and Cardiovascular Morbidity and Mortality: A Science Advisory From the American Heart Association.
Circulation.
2016; 134(13): e262–79. PubMed Abstract
| Publisher Full Text
5.
Donnelly JE, Blair SN, Jakicic JM, et al.:
American College of Sports Medicine Position Stand. Appropriate physical activity intervention strategies for weight loss and prevention of weight regain for adults.
Med Sci Sports Exerc.
2009; 41(2): 459–71. PubMed Abstract
| Publisher Full Text
6.
Tremblay A, Chaput JP:
Adaptive reduction in thermogenesis and resistance to lose fat in obese men.
Br J Nutr.
2009; 102(4): 488–92. PubMed Abstract
| Publisher Full Text
7.
Doucet E, St-Pierre S, Alméras N, et al.:
Evidence for the existence of adaptive thermogenesis during weight loss.
Br J Nutr.
2001; 85(6): 715–23. PubMed Abstract
| Publisher Full Text
8.
Redman LM, Heilbronn LK, Martin CK, et al.:
Metabolic and behavioral compensations in response to caloric restriction: implications for the maintenance of weight loss.
PLoS One.
2009; 4(2): e4377. PubMed Abstract
| Publisher Full Text
| Free Full Text
9.
Speakman JR, Selman C:
Physical activity and resting metabolic rate.
Proc Nutr Soc.
2003; 62(3): 621–34. PubMed Abstract
| Publisher Full Text
10.
Thomas DM, Bouchard C, Church T, et al.:
Why do individuals not lose more weight from an exercise intervention at a defined dose? An energy balance analysis.
Obes Rev.
2012; 13(10): 835–47. PubMed Abstract
| Publisher Full Text
| Free Full Text
11.
Byrne NM, Wood RE, Schutz Y, et al.:
Does metabolic compensation explain the majority of less-than-expected weight loss in obese adults during a short-term severe diet and exercise intervention?
Int J Obes (Lond).
2012; 36(11): 1472–8. PubMed Abstract
| Publisher Full Text
12.
Shaw K, Gennat H, O'Rourke P, et al.:
Exercise for overweight or obesity.
Cochrane Database Syst Rev.
2006; (4): CD003817. PubMed Abstract
| Publisher Full Text
13.
Jennings AE, Alberga A, Sigal RJ, et al.:
The effect of exercise training on resting metabolic rate in type 2 diabetes mellitus.
Med Sci Sports Exerc.
2009; 41(8): 1558–65. PubMed Abstract
| Publisher Full Text
14.
Amati F, Dubé JJ, Shay C, et al.:
Separate and combined effects of exercise training and weight loss on exercise efficiency and substrate oxidation.
J Appl Physiol (1985).
2008; 105(3): 825–31. PubMed Abstract
| Publisher Full Text
| Free Full Text
15.
Maclean PS, Bergouignan A, Cornier MA, et al.:
Biology's response to dieting: the impetus for weight regain.
Am J Physiol Regul Integr Comp Physiol.
2011; 301(3): R581–600. PubMed Abstract
| Publisher Full Text
| Free Full Text
16.
Garland T Jr, Schutz H, Chappell MA, et al.:
The biological control of voluntary exercise, spontaneous physical activity and daily energy expenditure in relation to obesity: human and rodent perspectives.
J Exp Biol.
2011; 214(Pt 2): 206–29. PubMed Abstract
| Publisher Full Text
| Free Full Text
18.
Levine JA, Eberhardt NL, Jensen MD:
Role of nonexercise activity thermogenesis in resistance to fat gain in humans.
Science.
1999; 283(5399): 212–4. PubMed Abstract
| Publisher Full Text
20.
Teich T, Dunford EC, Porras DP, et al.:
Glucocorticoid antagonism limits adiposity rebound and glucose intolerance in young male rats following the cessation of daily exercise and caloric restriction.
Am J Physiol Endocrinol Metab.
2016; 311(1): E56–68. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
21.
Lark DS, Fisher-Wellman KH, Neufer PD:
High-fat load: mechanism(s) of insulin resistance in skeletal muscle.
Int J Obes Suppl.
2012; 2(Suppl 2): S31–S36. PubMed Abstract
| Publisher Full Text
| Free Full Text
22.
Hesselink MK, Schrauwen-Hinderling V, Schrauwen P:
Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus.
Nat Rev Endocrinol.
2016; 12(11): 633–45. PubMed Abstract
| Publisher Full Text
23.
Theurey P, Rieusset J:
Mitochondria-Associated Membranes Response to Nutrient Availability and Role in Metabolic Diseases.
Trends Endocrinol Metab.
2017; 28(1): 32–45. PubMed Abstract
| Publisher Full Text
26.
Koves TR, Ussher JR, Noland RC, et al.:
Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance.
Cell Metab.
2008; 7(1): 45–56. PubMed Abstract
| Publisher Full Text
27.
Adams SH, Hoppel CL, Lok KH, et al.:
Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women.
J Nutr.
2009; 139(6): 1073–81. PubMed Abstract
| Publisher Full Text
| Free Full Text
28.
Noland RC, Koves TR, Seiler SE, et al.:
Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control.
J Biol Chem.
2009; 284(34): 22840–52. PubMed Abstract
| Publisher Full Text
| Free Full Text
29.
Hoehn KL, Salmon AB, Hohnen-Behrens C, et al.:
Insulin resistance is a cellular antioxidant defense mechanism.
Proc Natl Acad Sci U S A.
2009; 106(42): 17787–92. PubMed Abstract
| Publisher Full Text
| Free Full Text
30.
Muoio DM, Noland RC, Kovalik JP, et al.:
Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility.
Cell Metab.
2012; 15(5): 764–77. PubMed Abstract
| Publisher Full Text
| Free Full Text
31.
Gómez-Valadés AG, Méndez-Lucas A, Vidal-Alabró A, et al.:
Pck1 gene silencing in the liver improves glycemia control, insulin sensitivity, and dyslipidemia in db/db mice.
Diabetes.
2008; 57(8): 2199–210. PubMed Abstract
| Publisher Full Text
| Free Full Text
33.
Garbow JR, Doherty JM, Schugar RC, et al.:
Hepatic steatosis, inflammation, and ER stress in mice maintained long term on a very low-carbohydrate ketogenic diet.
Am J Physiol Gastrointest Liver Physiol.
2011; 300(6): G956–67. PubMed Abstract
| Publisher Full Text
| Free Full Text
36.
Fisher-Wellman KH, Neufer PD:
Linking mitochondrial bioenergetics to insulin resistance via redox biology.
Trends Endocrinol Metab.
2012; 23(3): 142–53. PubMed Abstract
| Publisher Full Text
| Free Full Text
39.
Kane DA, Anderson EJ, Price JW 3rd, et al.:
Metformin selectively attenuates mitochondrial H2O2 emission without affecting respiratory capacity in skeletal muscle of obese rats.
Free Radic Biol Med.
2010; 49(6): 1082–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
40.
Lark DS, Kang L, Lustig ME, et al.:
Enhanced mitochondrial superoxide scavenging does not improve muscle insulin action in the high fat-fed mouse.
PLoS One.
2015; 10(5): e0126732. PubMed Abstract
| Publisher Full Text
| Free Full Text
41.
Paglialunga S, van Bree B, Bosma M, et al.:
Targeting of mitochondrial reactive oxygen species production does not avert lipid-induced insulin resistance in muscle tissue from mice.
Diabetologia.
2012; 55(10): 2759–68. PubMed Abstract
| Publisher Full Text
42.
Anderson EJ, Yamazaki H, Neufer PD:
Induction of endogenous uncoupling protein 3 suppresses mitochondrial oxidant emission during fatty acid-supported respiration.
J Biol Chem.
2007; 282(43): 31257–66. PubMed Abstract
| Publisher Full Text
43.
Lark DS, Torres MJ, Lin CT, et al.:
Direct real-time quantification of mitochondrial oxidative phosphorylation efficiency in permeabilized skeletal muscle myofibers.
Am J Physiol Cell Physiol.
2016; 311(2): C239–45. PubMed Abstract
| Publisher Full Text
| Free Full Text
45.
Ost M, Keipert S, Klaus S:
Targeted mitochondrial uncoupling beyond UCP1 - The fine line between death and metabolic health.
Biochimie.
2017; 134: 77–85. PubMed Abstract
| Publisher Full Text
51.
Lackey DE, Olefsky JM:
Regulation of metabolism by the innate immune system.
Nat Rev Endocrinol.
2016; 12(1): 15–28. PubMed Abstract
| Publisher Full Text
52.
Hotamisligil GS, Shargill NS, Spiegelman BM:
Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance.
Science.
1993; 259(5091): 87–91. PubMed Abstract
| Publisher Full Text
53.
Plomgaard P, Bouzakri K, Krogh-Madsen R, et al.:
Tumor necrosis factor-alpha induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation.
Diabetes.
2005; 54(10): 2939–45. PubMed Abstract
| Publisher Full Text
54.
Lang CH, Dobrescu C, Bagby GJ:
Tumor necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose output.
Endocrinology.
1992; 130(1): 43–52. PubMed Abstract
| Publisher Full Text
55.
Kang L, Ayala JE, Lee-Young RS, et al.:
Diet-induced muscle insulin resistance is associated with extracellular matrix remodeling and interaction with integrin alpha2beta1 in mice.
Diabetes.
2011; 60(2): 416–26. PubMed Abstract
| Publisher Full Text
| Free Full Text
57.
Crewe C, An YA, Scherer PE:
The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis.
J Clin Invest.
2017; 127(1): 74–82. PubMed Abstract
| Publisher Full Text
| Free Full Text
59.
Spencer M, Yao-Borengasser A, Unal R, et al.:
Adipose tissue macrophages in insulin-resistant subjects are associated with collagen VI and fibrosis and demonstrate alternative activation.
Am J Physiol Endocrinol Metab.
2010; 299(6): E1016–27. PubMed Abstract
| Publisher Full Text
| Free Full Text
60.
Dankel SN, Svärd J, Matthä S, et al.:
COL6A3 expression in adipocytes associates with insulin resistance and depends on PPARγ and adipocyte size.
Obesity (Silver Spring).
2014; 22(8): 1807–13. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
63.
Khan T, Muise ES, Iyengar P, et al.:
Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI.
Mol Cell Biol.
2009; 29(6): 1575–91. PubMed Abstract
| Publisher Full Text
| Free Full Text
64.
Huber J, Löffler M, Bilban M, et al.:
Prevention of high-fat diet-induced adipose tissue remodeling in obese diabetic mice by n-3 polyunsaturated fatty acids.
Int J Obes (Lond).
2007; 31(6): 1004–13. PubMed Abstract
| Publisher Full Text
65.
Kiefer FW, Zeyda M, Todoric J, et al.:
Osteopontin expression in human and murine obesity: extensive local up-regulation in adipose tissue but minimal systemic alterations.
Endocrinology.
2008; 149(3): 1350–7. PubMed Abstract
| Publisher Full Text
66.
Nomiyama T, Perez-Tilve D, Ogawa D, et al.:
Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice.
J Clin Invest.
2007; 117(10): 2877–88. PubMed Abstract
| Publisher Full Text
| Free Full Text
67.
Han CY, Subramanian S, Chan CK, et al.:
Adipocyte-derived serum amyloid A3 and hyaluronan play a role in monocyte recruitment and adhesion.
Diabetes.
2007; 56(9): 2260–73. PubMed Abstract
| Publisher Full Text
68.
Varma V, Yao-Borengasser A, Bodles AM, et al.:
Thrombospondin-1 is an adipokine associated with obesity, adipose inflammation, and insulin resistance.
Diabetes.
2008; 57(2): 432–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
69.
Inoue M, Jiang Y, Barnes RH 2nd, et al.:
Thrombospondin 1 mediates high-fat diet-induced muscle fibrosis and insulin resistance in male mice.
Endocrinology.
2013; 154(12): 4548–59. PubMed Abstract
| Publisher Full Text
| Free Full Text
72.
Berria R, Wang L, Richardson DK, et al.:
Increased collagen content in insulin-resistant skeletal muscle.
Am J Physiol Endocrinol Metab.
2006; 290(3): E560–5. PubMed Abstract
| Publisher Full Text
76.
Kang L, Mayes WH, James FD, et al.:
Matrix metalloproteinase 9 opposes diet-induced muscle insulin resistance in mice.
Diabetologia.
2014; 57(3): 603–13. PubMed Abstract
| Publisher Full Text
| Free Full Text
78.
Ekstedt M, Franzén LE, Mathiesen UL, et al.:
Long-term follow-up of patients with NAFLD and elevated liver enzymes.
Hepatology.
2006; 44(4): 865–73. PubMed Abstract
| Publisher Full Text
79.
McCullough AJ:
The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease.
Clin Liver Dis.
2004; 8(3): 521–33, viii. PubMed Abstract
| Publisher Full Text
80.
Kohli R, Kirby M, Xanthakos SA, et al.:
High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis.
Hepatology.
2010; 52(3): 934–44. PubMed Abstract
| Publisher Full Text
| Free Full Text
83.
Gattazzo F, Urciuolo A, Bonaldo P:
Extracellular matrix: a dynamic microenvironment for stem cell niche.
Biochim Biophys Acta.
2014; 1840(8): 2506–19. PubMed Abstract
| Publisher Full Text
| Free Full Text
84.
Ringer P, Colo G, Fässler R, et al.:
Sensing the mechano-chemical properties of the extracellular matrix.
Matrix Biol.
2017; pii: S0945-053X(17)30018-5. PubMed Abstract
| Publisher Full Text
85.
Kim SH, Turnbull J, Guimond S:
Extracellular matrix and cell signalling: the dynamic cooperation of integrin, proteoglycan and growth factor receptor.
J Endocrinol.
2011; 209: 139–51. PubMed Abstract
| Publisher Full Text
87.
Schwander M, Shirasaki R, Pfaff SL, et al.:
Beta1 integrins in muscle, but not in motor neurons, are required for skeletal muscle innervation.
J Neurosci.
2004; 24(37): 8181–91. PubMed Abstract
| Publisher Full Text
88.
Schwander M, Leu M, Stumm M, et al.:
Beta1 integrins regulate myoblast fusion and sarcomere assembly.
Dev Cell.
2003; 4(5): 673–85. PubMed Abstract
| Publisher Full Text
89.
Liadaki K, Casar JC, Wessen M, et al.:
β4 integrin marks interstitial myogenic progenitor cells in adult murine skeletal muscle.
J Histochem Cytochem.
2012; 60(1): 31–44. PubMed Abstract
| Publisher Full Text
| Free Full Text
90.
Zong H, Bastie CC, Xu J, et al.:
Insulin resistance in striated muscle-specific integrin receptor beta1-deficient mice.
J Biol Chem.
2009; 284(7): 4679–88. PubMed Abstract
| Publisher Full Text
| Free Full Text
91.
Kang L, Mokshagundam S, Reuter B, et al.:
Integrin-Linked Kinase in Muscle Is Necessary for the Development of Insulin Resistance in Diet-Induced Obese Mice.
Diabetes.
2016; 65(6): 1590–600. PubMed Abstract
| Publisher Full Text
| Free Full Text
92.
Williams AS, Trefts E, Lantier L, et al.:
Integrin-Linked Kinase Is Necessary for the Development of Diet-Induced Hepatic Insulin Resistance.
Diabetes.
2017; 66(2): 325–34. PubMed Abstract
| Publisher Full Text
| Free Full Text
93.
Dobreva I, Fielding A, Foster LJ, et al.:
Mapping the integrin-linked kinase interactome using SILAC.
J Proteome Res.
2008; 7(4): 1740–9. PubMed Abstract
| Publisher Full Text
95.
Tu Y, Liang L, Frank SJ, et al.:
Src homology 3 domain-dependent interaction of Nck-2 with insulin receptor substrate-1.
Biochem J.
2001; 354(Pt 2): 315–22. PubMed Abstract
| Publisher Full Text
| Free Full Text
97.
Xu Z, Fukuda T, Li Y, et al.:
Molecular dissection of PINCH-1 reveals a mechanism of coupling and uncoupling of cell shape modulation and survival.
J Biol Chem.
2005; 280(30): 27631–7. PubMed Abstract
| Publisher Full Text
98.
Nikolopoulos SN, Turner CE:
Actopaxin, a new focal adhesion protein that binds paxillin LD motifs and actin and regulates cell adhesion.
J Cell Biol.
2000; 151(7): 1435–48. PubMed Abstract
| Publisher Full Text
| Free Full Text
100.
Pignatelli J, LaLonde SE, LaLonde DP, et al.:
Actopaxin (α-parvin) phosphorylation is required for matrix degradation and cancer cell invasion.
J Biol Chem.
2012; 287(44): 37309–20. PubMed Abstract
| Publisher Full Text
| Free Full Text
102.
Sylow L, Jensen TE, Kleinert M, et al.:
Rac1 signaling is required for insulin-stimulated glucose uptake and is dysregulated in insulin-resistant murine and human skeletal muscle.
Diabetes.
2013; 62(6): 1865–75. PubMed Abstract
| Publisher Full Text
| Free Full Text
104.
Chua BT, Volbracht C, Tan KO, et al.:
Mitochondrial translocation of cofilin is an early step in apoptosis induction.
Nat Cell Biol.
2003; 5(12): 1083–9. PubMed Abstract
| Publisher Full Text
1
Department of Molecular Physiology & Biophysics, Vanderbilt University School of Medicine, Nashville, TN, USA 2
Vanderbilt Mouse Metabolic Phenotyping Center, Vanderbilt University School of Medicine, Nashville, TN, USA
The author(s) declared that this work was funded by National Institutes of Health (grants DK054902, DK050277, DK059637), NIDDK Mouse Metabolic Phenotyping Centers MICROMouse Program (grant 15GRU2558) and American Heart Association (grant 16POST29910001).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Lark DS and Wasserman DH. Meta-fibrosis links positive energy balance and mitochondrial metabolism to insulin resistance [version 1; peer review: 3 approved]. F1000Research 2017, 6(F1000 Faculty Rev):1758 (https://doi.org/10.12688/f1000research.11653.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
NOTE: it is important to ensure the information in square brackets after the title is included in this citation.
Reviewer Report27 Sep 2017
Jane Shearer,
Department of Biochemistry & Molecular Biology, Faculty of Medicine, University of Calgary, Calgary, Alberta T2N1N4, Canada; Faculty of Kinesiology, University of Calgary, Calgary, Alberta T2N1N4, Canada
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles

Comments on this article Comments (0)