1Genomics Laboratories, Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, USA 2Schulze Center for Novel Therapeutics, Division of Oncology Research, Department of Oncology, Mayo Clinic, Rochester, USA 3Division of Research, Department of Surgery, Medical College of Wisconsin, Milwaukee, USA
Meghan M Kozub
Roles:
Conceptualization,
Writing – Original Draft Preparation
Ryan M Carr
Roles:
Conceptualization,
Writing – Original Draft Preparation,
Writing – Review & Editing
Gwen L Lomberk
Roles:
Conceptualization,
Writing – Original Draft Preparation
Martin E Fernandez-Zapico
Roles:
Conceptualization,
Resources,
Writing – Original Draft Preparation,
Writing – Review & Editing
OPEN PEER REVIEW
REVIEWER STATUS
Abstract
Histone-modifying enzymes play a critical role in chromatin remodeling and are essential for influencing several genome processes such as gene expression and DNA repair, replication, and recombination. The discovery of lysine-specific demethylase 1 (LSD1), the first identified histone demethylase, dramatically revolutionized research in the field of epigenetics. LSD1 plays a pivotal role in a wide range of biological operations, including development, cellular differentiation, embryonic pluripotency, and disease (for example, cancer). This mini-review focuses on the role of LSD1 in chromatin regulatory complexes, its involvement in epigenetic changes throughout development, and its importance in physiological and pathological processes.
Keywords
LSD1, KDM1a, histone demethylation, epigenetics, cancer
Corresponding author:
Martin E Fernandez-Zapico
Competing interests:
No competing interests were disclosed.
Grant information:
This work was supported by National Institutes of Health/National Cancer Institute CA136526, Mayo Clinic Pancreatic SPORE P50 CA102701, and Mayo Clinic Center for Cell Signaling in Gastroenterology P30 DK84567 to MEF-Z.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Within the nuclei of all eukaryotic cells, DNA is highly compacted via interactions with histones and numerous other proteins to form chromatin. Histones are involved with supercoiling of DNA and are subjected to post-translation modifications (PTMs)1. Diverse covalent modifications to histones control the structure and dynamics of chromatin and regulate access to DNA, ultimately altering gene expression. Multiple biochemical moieties can be covalently added to specific amino acids on the N-terminus of the histone, or “histone tail”1,2. The sequence of histone tail modifications, or “histone code”, influences transcription and other processes, including DNA repair, replication, and recombination3.
Histone PTMs were historically thought to be irreversible until the discovery of enzymes catalyzing the addition and removal of methyl groups to lysine and arginine residues on histone tails4. Two evolutionarily conserved families of histone demethylases that recognize H3K4me as a substrate have been identified: lysine-specific demethylases (LSDs) and Jumonji C demethylases (JMJCs)5–7. LSD enzymes demethylate mono- and di-methyl groups of lysine residues and some non-histone targets. JMJCs belong to the dioxygenase superfamily involved in deoxygenation reactions dependent on ferrous iron and α-ketoglutarate allowing demethylation of trimethylated lysine residues6,7. Lysine-specific histone demethylase I (LSD1) was first described in 2004, inspiring the hypothesis that histone modification is a highly dynamic process8.
This review focuses on LSD1 (also known as KDM1a) and its role in various physiological and pathological processes. LSD1 acts on histone H3 as a transcription co-repressor through demethylation of lysine 4 (H3K4) or as a transcription co-activator through demethylation of lysine 9 (H3K9)7,9–11. The enzyme is essential in the control of wide-ranging biological processes, including cell proliferation12, chromosome segregation13, hematopoiesis14, spermatogenesis15, adipogenesis16, stem cell pluripotency17, and embryonic development18. LSD1 can act as an oncogene, and its overexpression promotes cancer cell proliferation, migration, and invasion10,11.
LSD1 complex with regulatory proteins to facilitate histone demethylation
LSD1 consists of three structural domains: N-terminal SWIRM domain, C-terminal flavin adenine dinucleotide (FAD)-binding amine oxidase domain, and the tower domain. The SWIRM domain consists of proteins Swi3, Rsc8, and Moira. Through hydrophobic interactions, the FAD domain closely associated with SWIRM, forming a spherical core. Extending from the core is the tower domain forming an elongated helix-turn-helix motif.
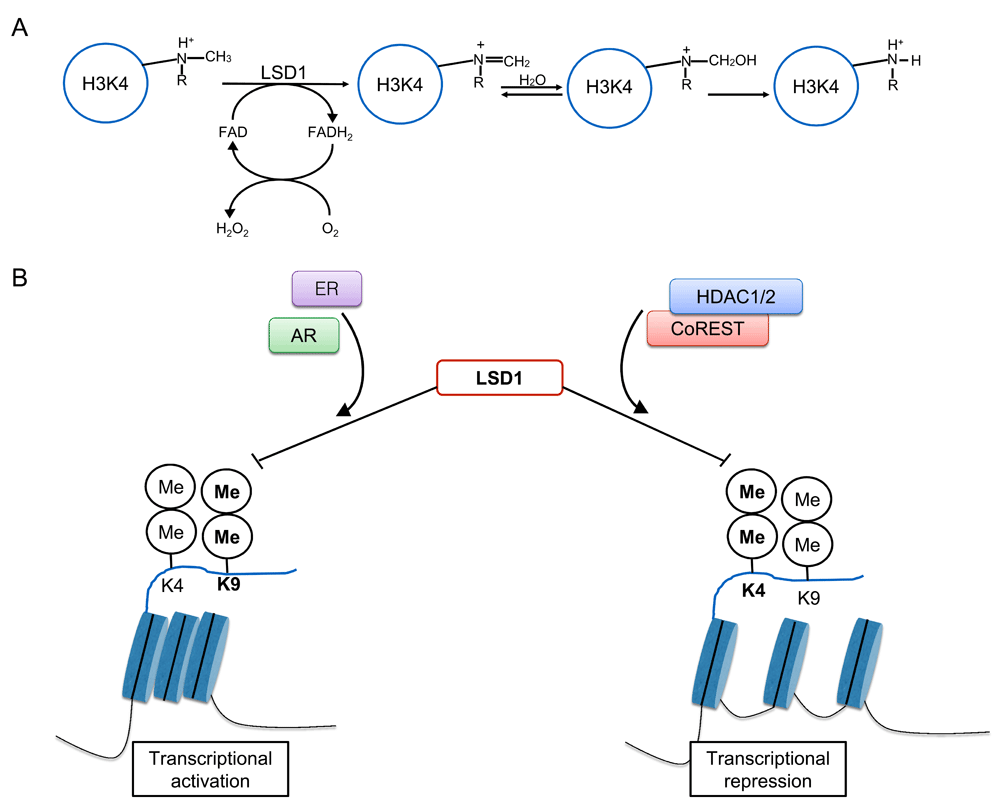
Frequently, LSD1 is found to be associated with a transcriptional co-repressor protein (CoREST) and histone deacetylase (HDAC) 1/2 to form a complex. Interaction with CoREST is necessary for LSD1 H3K4 demethylation19–22. Association of the FAD domain with CoREST-histone ternary complex results in conformational change, permitting association with the N-terminal H3 tail23,24. With H3K4 in close propinquity to the FAD domain, oxidative demethylation results in increased affinity for LSD123,25–27. LSD1 requires the first 20 N-terminal amino acids of the histone tail for substrate recognition and interaction28,29. The specific amino-acid sequence allows LSD1 to sense the epigenetic messages encoded within histone tail and efficiently carry out demethylation. The presence of other epigenetic marks on H3 has the potential to affect the enzymatic activity of LSD1, suggesting a regulatory role for the H3 tail bereft of all other epigenetic modifications upon LSD1 activity29,30.
Within the aforementioned complex, LSD1 demethylates mono- and di-methylated H3K47,25,30,31. Earlier studies identified REST as a long-term repressor of neuronal genes in non-neuronal cells19,32–34. This was determined to be mediated through the recruitment of the LSD1-CoREST-HDAC complex, thus allowing lysine deacetylation of H3 and H4 in addition to demethylation of H3K435. In other investigations, RNA interference (RNAi)-mediated knockdown of LSD1 resulted in H3K4 methylation mark recurrence in proximity to REST target promoters, confirming this regulatory function30. While methylation of H3K4 activates transcription, demethylation by LSD1 confers transcriptional repression32. LSD1 also serves as a transcriptional activator. For example, androgen receptor (AR) activation of its target genes is dependent upon LSD1-mediated H3K9 demethylation21. Following hormone treatment, AR and LSD1 co-localize to promoters, resulting in H3K9 demethylation without changing H3K4 methylation status9. As expected, LSD1 knockdown resulted in reduced activation of AR-responsive promoters7,35. Taken together, the above findings showed the potential of LSD1 as a transcriptional repressor or activator through dynamic and selective H3K4 demethylation and H3K9 demethylation, respectively (Figure 1).
Figure 1. LSD1, through histone 3 demethylation, has dual functions as both a transcriptional activator and repressor.
As an illustration of this, LSD1 (lysine-specific demethylase 1) can be associated with activated estrogen receptors (ERs) or androgen receptors (ARs) and promote demethylation at lysine 9 (K9) of the histone tail. This confers opening of heterochromatin, promoting transcriptional activation. Alternatively, LSD1 can complex with CoREST and histone deacetylase 1/2 (HDAC1/2). This association confers more specificity for methylated lysine 4 (K4), resulting in its demethylation. This promotes heterochromatin formation and transcriptional repression.
LSD1 contributes to broad, dynamic gene regulation through histone demethylation
Under physiological conditions, LSD1 has been found to have several functions ranging from regulation of hormone receptor–mediated transcription, appropriate hematopoietic stem cell differentiation, and cell cycle control. Estrogen receptor (ER)-mediated transcription is driven by LSD1-mediated H3K9 demethylation at both the gene promoter and an upstream enhancer site. ER then takes part in bending chromatin to interact with RNA polymerase to promote transcription. LSD1 co-localizes to this DNA loop, and the process of demethylation results in formation of hydrogen peroxide, inducing local oxidative DNA damage, recruiting 8-oxoguanine-DNA glycosylase 1 and topoisomerase IIβ. These enzymes induce DNA conformational changes necessary for efficient promotion of gene expression36,37. Thus, LSD1 plays an important role in hormone receptor–mediated gene expression via histone demethylation and through DNA damage–induced chromatin remodeling.
In addition to driving hormone receptor–mediated gene expression, LSD1 plays a critical role in hematopoiesis. LSD1 is dynamically involved in hematopoietic differentiation through cooperation with growth factor–independent (GFI) proteins. GFI proteins promote expression of lineage-specific genes, and LSD1 specifically interacts with GFI1B binding sites. In mouse models, RNAi depletion of LSD1 impairs both erythrocyte and megakaryocyte differentiation but activated spontaneous granulocyte differentiation. Therefore, actions of LSD1 may be lineage-specific38. However, alternative models have revealed competing findings that LSD1 is necessary for terminal differentiation of erythroid, megakaryocytic, and granulocytic lineages38–40. Although its specific role is not fully characterized, LSD1 was found to regulate promoters and enhancers of genes associated with hematopoietic stem cells and was critical for terminal differentiation of mature hematopoietic cells as LSD1 knockout resulted in severe pancytopenia41.
Finally, LSD1 biological functions are associated with the regulation of the methylation status of non-histone proteins. Studies demonstrate the relationship between retinoblastoma gene (RB1) and LSD1 in cell cycle control. Overexpression of RB1, the first identified tumor suppressor, causes cells to undergo arrest in the G1 phase of the cell cycle, and, as expected, abrogation of RB1 accelerates G1 transition42,43. Phosphorylation is a key mechanism by which RB1 is regulated. Dephosphorylation is mediated by myosin phosphatase, which promotes cell cycle arrest44. Interestingly, myosin phosphatase target subunit 1 (MYPT1) was identified as a novel substrate of LSD1. Methylation of MYPT1 is mediated by LSD1, preventing dephosphorylation of RB1 and ultimately promoting cell cycle progression12. Given these diverse functions, one can see how altered LSD1 activity could contribute significantly to normal homeostasis and pathology such as malignancy.
Given the aforementioned role of LSD1 in cell cycle regulation, one may hypothesize that it could serve as an oncogene in the context of malignant transformation. LSD1 was first found to be overexpressed in neuroblastoma, correlating with poor differentiation45. Overexpression of LSD1 has been documented in many solid tumors and is correlated with aggressive clinicopathological features and poor patient outcomes11,46–49. Both in vitro and in vivo models have demonstrated overexpression of LSD1 correlating with significant chromatin modifications and malignant transformation50,51. Both pharmacological inhibition and genetic depletion of LSD1 have been shown to inhibit cancer cell proliferation, differentiation, invasion, and metastasis in animal models7,40,52. Thus, LSD1 has been confirmed to be an important oncogenic driver, a potential biomarker indicative of poor prognosis, and a potential therapeutic target. There are several forms of malignancy that have been shown to have aberrant LSD1 activity.
First, LSD1 is critical in the process of terminal differentiation in hematopoiesis, and abnormal LSD1 activity is correlated with a variety of myeloproliferative disorders11,50,51,53. Many studies propose LSD1 as a prospective treatment target for acute myeloid leukemia (AML). AML is a heterogenous hematopoietic malignancy characterized by the accumulation of incompletely differentiated progenitor cells (blasts) in the bone marrow, causing suppression of normal hematopoiesis41. LSD1 is a required constituent of a mixed-lineage leukemia (MLL) super complex associated with active transcription sites54. Abrogation of LSD1 results in heightened rates of apoptosis and impaired leukemogenicity in an MLL-AF9 mouse model55. In acute promyelocytic leukemia, all-trans retinoic acid can induce differentiation of leukemic cells whereas AML is not responsive. However, inhibition of LSD1 activity in AML models results in increased H3K4me2 at myeloid differentiation–associated genes, resulting in increased responsiveness to all-trans retinoic acid40. These data support the importance of LSD1-mediated alteration of the leukemic epigenome in pathogenesis and demonstrate how the enzyme could serve as a therapeutic target in AML.
LSD1 is also dysregulated in solid tumors, including colorectal carcinoma. Increased activity in colon cancer is associated with increased metastatic potential56. Higher expression of LSD1 and low expression of CDH-1 (E-cadherin) in colorectal cancer were associated with higher tumor-node-metastasis staging and thus poorer prognosis57. Knockdown of LSD1 results in CDH-1 upregulation and confers reduced invasiveness. LSD1 regulates the CDH-1 promoter and demethylation of H3K4me2 causes downregulation of CDH-1 expression58.
Squamous cell carcinoma (SCC) is also associated with elevated LSD1 activity. The most common genetic aberration in SCC of the lung, esophagus, and oral cavity is amplification of Sox259–61. Sox2 encodes a transcription factor important in embryonic stem cells with the ability to reprogram somatic cells into induced pluripotent stem cells. Elevated LSD1 levels are associated with amplified Sox2 expression in lung cancer. Cells from these patients are particularly sensitive to the LSD1 inhibitor, CBB1007, whereas Sox2-negative cells are not62. Subsequent chromatin immunoprecipitation (ChIP) sequencing revealed that LSD1 binds to the Sox2 gene and is enriched in the transcriptional regulator region, a known distant enhancer for Sox2 expression in breast cancer. LSD1 inhibition revealed that its activity is required for Sox2 expression. Inactivation results in increased global H3K9me1/me2 and H3K27me3 with formation of bivalent chromatin domains within the regulatory regions of Sox2 and cell cycle regulatory genes, leading to suppression of gene expression63.
ER-negative breast cancer is a subtype of the common malignancy with relatively more rapid growth, loss of differentiation, and increased propensity for metastasis. Interestingly, LSD1 tends to be highly expressed in this form of breast cancer64. LSD1 and HDAC closely interact and control the growth of breast cancer through aberrant gene silencing65.
Upregulation of LSD1 promotes epithelial-to-mesenchymal transition
Aberrant LSD1 activity is extensively characterized in multiple cancers, but the mechanism by which it promotes cancer progression extends beyond suppression of cell cycle regulators. For example, through PTM of a notable non-histone protein, p53, LSD1 represses apoptosis. This is achieved through demethylation of K370me2. Whereas methylation at this site promotes association of p53 with co-activator 53BP1, LSD1 inhibits this interaction66. A more thoroughly studied role of LSD1 is in the epithelial-to-mesenchymal transition, a critical process in cancer progression. Snail and Slug are key molecular mediators of epithelial-to-mesenchymal transition through direct repression of epithelial markers such as CDH-1. This is achieved through the SNAG domain of Snail, structurally resembling the histone H3 tail, recruiting LSD1 to epithelial gene promoters with formation of the Snail-LSD1-CoREST complex with subsequent demethylation of H3K4me267,68. In the specific case of neuroblastoma, MYCN has been correlated with poor prognosis. This is related to the co-localization of LSD1 and MYCN at the promoter of a key suppressor of metastasis, N-Myc downstream-regulated gene 1 (NDRG1), inhibiting its expression. Thus, elevated LSD1 is associated with lower NDRG1 expression and poor prognosis69. Inhibition of enzymatic activity or abrogation of the SNAG-LSD1 interaction suppresses mesenchymal markers, decreasing cancer invasiveness70,71. Promoting transition to a mesenchymal phenotype is opposed by acetylation of LSD1 by males absent on the first (MOF). In fact, MOF expression correlates with favorable prognosis in cancer72.
In summary, there is an abundance of evidence for a role of LSD1 in the pathogenicity of a wide array of malignancies. Cancer is the consequence of complex and heterogeneous genetic alterations, and aberrant LSD1 activity can contribute to a malignant phenotype through extensive modifications of the epigenome. Therefore, therapeutic targeting of the demethylase may prove an effective strategy in reversing or attenuating more aggressive malignant phenotypes in many cancers.
Conclusions
This review has briefly summarized the current knowledge and research of LSD1, its expression patterns, recruitment mechanisms, chromatin remodeling, biochemical functions, molecular structure, and role in cancer. In the past several years, studies have elucidated the role that histone lysine demethylases play in epigenetic regulation. LSD1 was the first histone demethylase identified and catalyzes the oxidation of methylated H3K4 through an amine oxidation reaction. Furthermore, LSD1 can act on non-histone proteins. While LSD1 is critical in conferring the dynamic nature of epigenetic regulation through histone modification, imbalance in histone modification with excessive LSD1 activity is significantly associated with increased cellular growth and suppression of cell cycle regulatory proteins in a broad array of tissues. Thus, LSD1 represents a critical oncogene and potential therapeutic target. More is to be elucidated regarding the function of LSD1. Interestingly, Wang et al. reported that LSD1 inhibits the invasion of breast cancer cells in vitro but conversely suppresses breast cancer metastatic potential in vivo73. These data suggest not only that LSD1 is multifunctional but also that its functions may be highly context-dependent. More extensive studies into the effects of these contexts on LSD1 function will be important to fully understand its role in cancer. A significant amount of research has been devoted to the development of a wide range of epigenetic therapies. Numerous questions still must be answered regarding LSD1 if effective therapeutic strategies are to be developed. For example, what are the functions and interactions of other LSD1 domains, or does LSD interact with methyltransferases, and how are these relationships regulated? In regard to the development of further therapies, emphasis should be placed on the development of highly specific drugs for demethylase subtypes to better direct the desired epigenetic effect of the drug. This would help specifically discern enzyme subtype mechanism. Despite these challenges, LSD1 is clearly important in normal cellular functions and malignancy.
Competing interests
The authors declare that they have no competing interests.
Grant information
This work was supported by National Institutes of Health/National Cancer Institute CA136526, Mayo Clinic Pancreatic SPORE P50 CA102701, and Mayo Clinic Center for Cell Signaling in Gastroenterology P30 DK84567 to MEF-Z.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We would like to thank Emily Porcher for her secretarial assistance. We would like to acknowledge the contributions of the authors of the excellent research studies and comprehensive reviews that were cited herein. We apologize to any authors whose work we omitted because of space limitations.
4.
Pedersen MT, Helin K:
Histone demethylases in development and disease.
Trends Cell Biol.
2010; 20(11): 662–71. PubMed Abstract
| Publisher Full Text
5.
Klose RJ, Kallin EM, Zhang Y:
JmjC-domain-containing proteins and histone demethylation.
Nat Rev Genet.
2006; 7(9): 715–27. PubMed Abstract
| Publisher Full Text
6.
Chen X, Hu Y, Zhou DX:
Epigenetic gene regulation by plant Jumonji group of histone demethylase.
Biochim Biophys Acta.
2011; 1809(8): 421–6. PubMed Abstract
| Publisher Full Text
8.
"KDM1A Lysine Demethylase 1A [Homo Sapiens (human)]". National Center for Biotechnology Information. U.S. National Library of Medicine, n.d. Web. 29 June 2016.
10.
Scoumanne A, Chen X:
The lysine-specific demethylase 1 is required for cell proliferation in both p53-dependent and -independent manners.
J Biol Chem.
2007; 282(21): 15471–5. PubMed Abstract
| Publisher Full Text
11.
Hayami S, Kelly JD, Cho HS, et al.:
Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers.
Int J Cancer.
2011; 128(3): 574–86. PubMed Abstract
| Publisher Full Text
12.
Cho HS, Suzuki T, Dohmae N, et al.:
Demethylation of RB regulator MYPT1 by histone demethylase LSD1 promotes cell cycle progression in cancer cells.
Cancer Res.
2011; 71(3): 655–60. PubMed Abstract
| Publisher Full Text
13.
Lv S, Bu W, Jiao H, et al.:
LSD1 is required for chromosome segregation during mitosis.
Eur J Cell Biol.
2010; 89(7): 557–63. PubMed Abstract
| Publisher Full Text
14.
Li Y, Deng C, Hu X, et al.:
Dynamic interaction between TAL1 oncoprotein and LSD1 regulates TAL1 function in hematopoiesis and leukemogenesis.
Oncogene.
2012; 31(48): 5007–18. PubMed Abstract
| Publisher Full Text
| Free Full Text
15.
Godmann M, Auger V, Ferraroni-Aguiar V, et al.:
Dynamic regulation of histone H3 methylation at lysine 4 in mammalian spermatogenesis.
Biol Reprod.
2007; 77(5): 754–64. PubMed Abstract
| Publisher Full Text
17.
Zhou H, Li W, Zhu S, et al.:
Conversion of mouse epiblast stem cells to an earlier pluripotency state by small molecules.
J Biol Chem.
2010; 285(39): 29676–80. PubMed Abstract
| Publisher Full Text
| Free Full Text
18.
Foster CT, Dovey OM, Lezina L, et al.:
Lysine-specific demethylase 1 regulates the embryonic transcriptome and CoREST stability.
Mol Cell Biol.
2010; 30(20): 4851–63. PubMed Abstract
| Publisher Full Text
| Free Full Text
19.
Ballas N, Battaglioli E, Atouf F, et al.:
Regulation of neuronal traits by a novel transcriptional complex.
Neuron.
2001; 31(3): 353–65. PubMed Abstract
| Publisher Full Text
20.
Shi Y, Sawada J, Sui G, et al.:
Coordinated histone modifications mediated by a CtBP co-repressor complex.
Nature.
2003; 422(6933): 735–8. PubMed Abstract
| Publisher Full Text
21.
Humphrey GW, Wang Y, Russanova VR, et al.:
Stable histone deacetylase complexes distinguished by the presence of SANT domain proteins CoREST/kiaa0071 and Mta-L1.
J Biol Chem.
2001; 276(9): 6817–24. PubMed Abstract
| Publisher Full Text
22.
You A, Tong JK, Grozinger CM, et al.:
CoREST is an integral component of the CoREST- human histone deacetylase complex.
Proc Natl Acad Sci U S A.
2001; 98(4): 1454–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
24.
Yang M, Culhane JC, Szewczuk LM, et al.:
Structural basis of histone demethylation by LSD1 revealed by suicide inactivation.
Nat Struct Mol Biol.
2007; 14(6): 535–9. PubMed Abstract
| Publisher Full Text
27.
Chen Y, Yang Y, Wang F, et al.:
Crystal structure of human histone lysine-specific demethylase 1 (LSD1).
Proc Natl Acad Sci U S A.
2006; 103(38): 13956–61. PubMed Abstract
| Publisher Full Text
| Free Full Text
28.
Forneris F, Binda C, Vanoni MA, et al.:
Human histone demethylase LSD1 reads the histone code.
J Biol Chem.
2005; 280(50): 41360–5. PubMed Abstract
| Publisher Full Text
29.
Forneris F, Binda C, Dall'Aglio A, et al.:
A highly specific mechanism of histone H3-K4 recognition by histone demethylase LSD1.
J Biol Chem.
2006; 281(46): 35289–95. PubMed Abstract
| Publisher Full Text
31.
Lee MG, Wynder C, Cooch N, et al.:
An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation.
Nature.
2005; 437(7057): 432–5. PubMed Abstract
| Publisher Full Text
34.
Ooi L, Wood IC:
Chromatin crosstalk in development and disease: lessons from REST.
Nat Rev Genet.
2007; 8(7): 544–54. PubMed Abstract
| Publisher Full Text
35.
Forneris F, Binda C, Vanoni MA, et al.:
Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process.
FEBS Lett.
2005; 579(10): 2203–7. PubMed Abstract
| Publisher Full Text
38.
Adamo A, Sesé B, Boue S, et al.:
LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells.
Nat Cell Biol.
2011; 13(6): 652–9. PubMed Abstract
| Publisher Full Text
39.
Lokken AA, Zeleznik-Le NJ:
Breaking the LSD1/KDM1A addiction: therapeutic targeting of the epigenetic modifier in AML.
Cancer Cell.
2012; 21(4): 451–3. PubMed Abstract
| Publisher Full Text
| Free Full Text
42.
Lee WH, Bookstein R, Hong F, et al.:
Human retinoblastoma susceptibility gene: cloning, identification, and sequence.
Science.
1987; 235(4794): 1394–9. PubMed Abstract
| Publisher Full Text
43.
Goodrich DW, Wang NP, Qian YW, et al.:
The retinoblastoma gene product regulates progression through the G1 phase of the cell cycle.
Cell.
1991; 67(2): 293–302. PubMed Abstract
| Publisher Full Text
44.
Classon M, Harlow E:
The retinoblastoma tumour suppressor in development and cancer.
Nat Rev Cancer.
2002; 2(12): 910–7. PubMed Abstract
| Publisher Full Text
45.
Schulte JH, Lim S, Schramm A, et al.:
Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy.
Cancer Res.
2009; 69(5): 2065–71. PubMed Abstract
| Publisher Full Text
47.
Magerl C, Ellinger J, Braunschweig T, et al.:
H3K4 dimethylation in hepatocellular carcinoma is rare compared with other hepatobiliary and gastrointestinal carcinomas and correlates with expression of the methylase Ash2 and the demethylase LSD1.
Hum Pathol.
2010; 41(2): 181–9. PubMed Abstract
| Publisher Full Text
48.
Lim S, Janzer A, Becker A, et al.:
Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology.
Carcinogenesis.
2010; 31(3): 512–20. PubMed Abstract
| Publisher Full Text
49.
Bennani-Baiti IM, Machado I, Llombart-Bosch A, et al.:
Lysine-specific demethylase 1 (LSD1/KDM1A/AOF2/BHC110) is expressed and is an epigenetic drug target in chondrosarcoma, Ewing's sarcoma, osteosarcoma, and rhabdomyosarcoma.
Hum Pathol.
2012; 43(8): 1300–7. PubMed Abstract
| Publisher Full Text
50.
Lv T, Yuan D, Miao X, et al.:
Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer.
PLoS One.
2012; 7(4): e35065. PubMed Abstract
| Publisher Full Text
| Free Full Text
51.
Kauffman EC, Robinson BD, Downes MJ, et al.:
Role of androgen receptor and associated lysine-demethylase coregulators, LSD1 and JMJD2A, in localized and advanced human bladder cancer.
Mol Carcinog.
2011; 50(12): 931–44. PubMed Abstract
| Publisher Full Text
| Free Full Text
52.
Huang Y, Greene E, Murray Stewart T, et al.:
Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes.
Proc Natl Acad Sci U S A.
2007; 104(19): 8023–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
53.
Kahl P, Gullotti L, Heukamp LC, et al.:
Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence.
Cancer Res.
2006; 66(23): 11341–7. PubMed Abstract
| Publisher Full Text
54.
Harris WJ, Huang X, Lynch JT, et al.:
The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells.
Cancer Cell.
2012; 21(4): 473–87. PubMed Abstract
| Publisher Full Text
55.
Mould DP, McGonagle AE, Wiseman DH, et al.:
Reversible inhibitors of LSD1 as therapeutic agents in acute myeloid leukemia: clinical significance and progress to date.
Med Res Rev.
2015; 35(3): 586–618. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
56.
Singh AB, Sharma A, Smith JJ, et al.:
Claudin-1 up-regulates the repressor ZEB-1 to inhibit E-cadherin expression in colon cancer cells.
Gastroenterology.
2011; 141(6): 2140–53. PubMed Abstract
| Publisher Full Text
| Free Full Text
57.
Jie D, Zhongmin Z, Guoqing L, et al.:
Positive expression of LSD1 and negative expression of E-cadherin correlate with metastasis and poor prognosis of colon cancer.
Dig Dis Sci.
2013; 58(6): 1581–9. PubMed Abstract
| Publisher Full Text
58.
Ding J, Zhang ZM, Xia Y, et al.:
LSD1-mediated epigenetic modification contributes to proliferation and metastasis of colon cancer.
Br J Cancer.
2013; 109(4): 994–1003. PubMed Abstract
| Publisher Full Text
| Free Full Text
59.
Bass AJ, Watanabe H, Mermel CH, et al.:
SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas.
Nat Genet.
2009; 41(11): 1238–42. PubMed Abstract
| Publisher Full Text
| Free Full Text
60.
Hussenet T, Dali S, Exinger J, et al.:
SOX2 is an oncogene activated by recurrent 3q26.3 amplifications in human lung squamous cell carcinomas.
PLoS One.
2010; 5(1): e8960. PubMed Abstract
| Publisher Full Text
| Free Full Text
61.
Maier S, Wilbertz T, Braun M, et al.:
SOX2 amplification is a common event in squamous cell carcinomas of different organ sites.
Hum Pathol.
2011; 42(8): 1078–88. PubMed Abstract
| Publisher Full Text
63.
Leis O, Eguiara A, Lopez-Arribillaga E, et al.:
Sox2 expression in breast tumours and activation in breast cancer stem cells.
Oncogene.
2012; 31(11): 1354–65. PubMed Abstract
| Publisher Full Text
64.
Cui X, Schiff R, Arpino G, et al.:
Biology of progesterone receptor loss in breast cancer and its implications for endocrine therapy.
J Clin Oncol.
2005; 23(30): 7721–35. PubMed Abstract
| Publisher Full Text
65.
Huang Y, Vasilatos SN, Boric L, et al.:
Inhibitors of histone demethylation and histone deacetylation cooperate in regulating gene expression and inhibiting growth in human breast cancer cells.
Breast Cancer Res Treat.
2012; 131(3): 777–89. PubMed Abstract
| Publisher Full Text
| Free Full Text
66.
Huang J, Sengupta R, Espejo AB, et al.:
p53 is regulated by the lysine demethylase LSD1.
Nature.
2007; 449(7158): 105–8. PubMed Abstract
| Publisher Full Text
67.
Lin T, Ponn A, Hu X, et al.:
Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition.
Oncogene.
2010; 29(35): 4896–904. PubMed Abstract
| Publisher Full Text
| Free Full Text
69.
Ambrosio S, Amente S, Saccà CD, et al.:
LSD1 mediates MYCN control of epithelial-mesenchymal transition through silencing of metastatic suppressor NDRG1 gene.
Oncotarget.
2017; 8(3): 3854–69. PubMed Abstract
| Publisher Full Text
| Free Full Text
73.
Wang Y, Zhang H, Chen Y, et al.:
LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer.
Cell.
2009; 138(4): 660–72. PubMed Abstract
| Publisher Full Text
1
Genomics Laboratories, Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, USA 2
Schulze Center for Novel Therapeutics, Division of Oncology Research, Department of Oncology, Mayo Clinic, Rochester, USA 3
Division of Research, Department of Surgery, Medical College of Wisconsin, Milwaukee, USA
Meghan M Kozub
Roles:
Conceptualization,
Writing – Original Draft Preparation
Ryan M Carr
Roles:
Conceptualization,
Writing – Original Draft Preparation,
Writing – Review & Editing
Gwen L Lomberk
Roles:
Conceptualization,
Writing – Original Draft Preparation
Martin E Fernandez-Zapico
Roles:
Conceptualization,
Resources,
Writing – Original Draft Preparation,
Writing – Review & Editing
This work was supported by National Institutes of Health/National Cancer Institute CA136526, Mayo Clinic Pancreatic SPORE P50 CA102701, and Mayo Clinic Center for Cell Signaling in Gastroenterology P30 DK84567 to MEF-Z.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Kozub MM, Carr RM, Lomberk GL and Fernandez-Zapico ME. LSD1, a double-edged sword, confers dynamic chromatin regulation but commonly promotes aberrant cell growth [version 1; peer review: 2 approved]. F1000Research 2017, 6(F1000 Faculty Rev):2016 (https://doi.org/10.12688/f1000research.12169.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)