Both actin and microtubules are major cytoskeletal elements in eukaryotic cells that participate in many cellular processes, including cell division and motility, vesicle and organelle movement, and the maintenance of cell shape. Inside its host cell, the human pathogen Chlamydia trachomatis manipulates the cytoskeleton to promote its survival and enhance its pathogenicity. In particular, Chlamydia induces the drastic rearrangement of both actin and microtubules, which is vital for its entry, inclusion structure and development, and host cell exit. As significant progress in Chlamydia genetics has greatly enhanced our understanding of how this pathogen co-opts the host cytoskeleton, we will discuss the machinery used by Chlamydia to coordinate the reorganization of actin and microtubules.
Corresponding author:
Fabienne Paumet
Competing interests:
No competing interests were disclosed.
Grant information:
This work was supported by National Institutes of Health grant AI116983 to Fabienne Paumet
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The Chlamydiaceae constitute a family of obligate intracellular bacteria that encompasses numerous species. With ~92 million new cases/year worldwide, Chlamydia trachomatis is the most frequent cause of bacterial sexually transmitted infections and is the leading cause of preventable infectious blindness called trachoma1–3. Trachoma is a significant problem in the developing world, where access to healthcare is limited and antibiotics are scarce. Chlamydia infections are also associated with chronic diseases and increased risk for cervical cancer4,5, making this infection a significant socioeconomic and medical burden in both developed and developing countries.
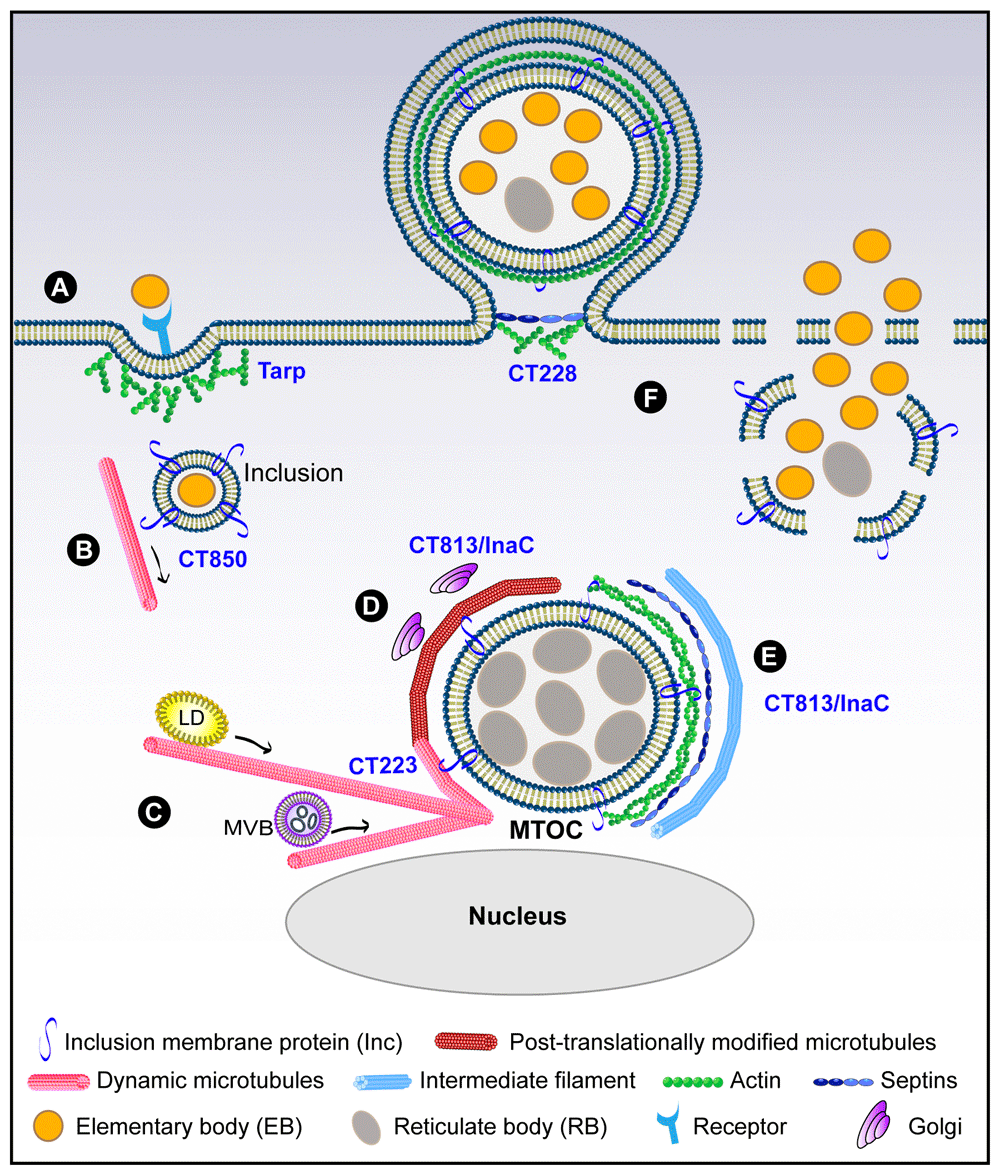
Chlamydiae exhibit a unique biphasic life cycle, cycling between a metabolically inactive but infectious small elementary body (EB ~200 nm) and a noninfectious metabolically active and dividing large reticulate body (RB ~800 nm)6. Chlamydia spends the majority of its life as an intracellular RB. Soon after invasion, EBs differentiate into RBs and replicate within a membrane-bound compartment called an “inclusion”. This obligate intracellular lifestyle required Chlamydia to develop an effective strategy to manipulate host cell pathways in order to ensure its survival and replication. Among the many pathways that Chlamydia co-opts, a common and intriguing target for all Chlamydia species has emerged: the host cytoskeleton7. We will discuss the recent advances concerning the role played by the host cytoskeleton in the growth and structural maintenance of the chlamydial inclusion (see Figure 1).
Figure 1. Reorganization of the host cytoskeleton during Chlamydia trachomatis infection.
(A) Entry during which a translocated actin-recruiting phosphoprotein (Tarp) induces actin polymerization; (B) transport of the nascent inclusion to the microtubule-organizing center (MTOC) using CT850; (C) formation of microtubule cages around the inclusion, in which CT223 is likely involved, and microtubule-dependent movement of lipid droplets (LDs) and multi-vesicular bodies (MVBs) towards the inclusion; (D) post-translational modifications of microtubule cages and positioning of Golgi mini-stacks around the inclusion controlled by CT813/InaC; (E) structural scaffolds of actin, septins, and intermediate filaments reinforce the growing inclusion membrane in a CT813-dependent manner; (F) Chlamydia exits the host cell using CT228-dependent extrusion (left) or through cell lysis (right).
Chlamydia trachomatis recruits actin to enter its host cell and uses microtubules to travel to the microtubule-organizing center
Because of their obligate intracellular nature, Chlamydiae have evolved very efficient ways to enter eukaryotic cells (Figure 1A). During infection, EBs attach to the host cell surface via a relatively weak electrostatic interaction with heparan sulfate moieties8. Then a stronger and more specific binding to a cellular receptor takes place, during which the majority of the EBs are internalized via an actin-dependent event9. While a number of receptors have been identified, including PDGFRβ, β1-integrin, and Ephrin A2, depletion of a single receptor is not sufficient to block entry, suggesting that Chlamydia utilizes more than one receptor to invade its host cell10,11.
To promote entry into non-phagocytic epithelial cells, C. trachomatis delivers a translocated actin-recruiting phosphoprotein (Tarp, also known as CT456)12–14 into the host cytoplasm (Figure 1A). In a phosphorylation-dependent manner, Tarp recruits guanine nucleotide exchange factors that activate Rac1, a member of the Rho family of GTPases9,10,13,15. Whereas Chlamydia caviae uses both Rac1 and Cdc42 to promote its entry, C. trachomatis recruits only Rac1, and not Cdc42 or RhoA16,17. Hijacking only one Rho GTPase family member is unique to C. trachomatis, as other intracellular bacteria including Salmonella and Shigella usually use multiple isoforms such as RhoA and Cdc4218–20. In addition to its Rac1 recruitment function, Tarp also directly binds to actin monomers and nucleates new unbranched actin filaments21,22. These linear filaments are then branched via the host Arp2/3 complex, which is activated by the Rac1 signaling pathway9,13. Thus, Tarp functions as both an actin nucleator and a signaling platform to locally remodel the actin cytoskeleton and promote Chlamydia invasion.
Nascent C. trachomatis-containing inclusions then use the minus-end-directed microtubule motor dynein to move from the cell periphery to the microtubule-organizing center (MTOC), where the inclusion resides for the duration of the life cycle23 (Figure 1B). This is a pathogen-driven event in which the inclusion protein CT850 is involved through its interaction with the dynein light chain DYNLT124. At the MTOC, Src family kinases control the tight association between inclusions and centrosomes25. Additional inclusion proteins including IncB, CT101, and CT222 are concentrated at these contact points between inclusions and centrosomes, suggesting their potential contribution to the transport of the inclusion24. In fact, during Chlamydia psittaci infection, IncB has been shown to interact with Snapin, which also binds dynein, thus connecting the inclusion to the microtubule network26. The association of inclusions with the MTOC is a common characteristic for a number of Chlamydia species, suggesting that this event is essential for Chlamydia’s life cycle. One possibility is that the MTOC brings host organelles and chlamydial inclusions in close proximity, thus facilitating the transfer of nutrients and lipids from the host to the inclusion. Additionally, the clustering of the inclusions at the MTOC is necessary for the homotypic fusion of inclusions to take place during C. trachomatis infection, as the dissociation of the inclusions from the MTOC inhibits this fusion event27. Homotypic fusion is critical for C. trachomatis pathogenicity, as non-fusing mutants grow significantly slower than their wild-type counterparts28,29. In particular, C. trachomatis strains that do not undergo homotypic fusion are also replication-defective and cause significantly milder disease in humans28–30. Given the importance of microtubule-based transport of the inclusion in Chlamydia development, additional unidentified Chlamydia effectors are likely involved in this process.
Chlamydia creates microtubule cages to support the development of its inclusion
Around 12 hours post-infection (PI), microtubules likely assemble around the inclusion under the control of the chlamydial effector CT223/IPAM, which has been shown to alter microtubule organization through the host centrosomal protein of 170 kDa (CEP170) in transfected cells31 (Figure 1C). There, microtubules encasing the inclusion are stabilized, which allows them to undergo post-translational modifications (PTMs), including detyrosination and acetylation32,33 (Figure 1D). These PTMs influence microtubule structure and depolymerization rates34 and have been implicated in the relocation of the Golgi apparatus around the chlamydial inclusion32,33. In most cells, dynamic microtubules have a half-life of about 5–10 minutes, while modified microtubules can persist for hours35, suggesting that Chlamydia uses these PTM microtubules to establish a stable long-term relationship with its host. Detyrosination is the best-characterized modification and involves the removal of the carboxy-terminal tyrosine from α-tubulin by tubulin carboxypeptidase, thus exposing a glutamic acid as the new C-terminus36.
Interestingly, stable microtubules are involved in the repositioning of organelles. During infection, the Golgi is a major source of host lipids, including sphingomyelin and cholesterol37–40. To enhance access to these lipids, Chlamydia induces the fragmentation of the Golgi into mini-stacks, which are then recruited around the inclusion in a microtubule-dependent manner32,41 (Figure 1D). In addition to controlling Golgi positioning, detyrosinated microtubules are involved in other trafficking events, including the recycling of endocytosed transferrin42 and the dispersal of lipid droplets43, which are also co-opted by Chlamydia39,44. Lipid droplets and multi-vesicular bodies are redirected towards the chlamydial inclusion along microtubules to provide fatty acids, which are important for Chlamydia replication39,40,44,45 (Figure 1C). The endoplasmic reticulum (ER) has also been associated with stable microtubules, in particular acetylated microtubules, along which ER tubules slide46. Chlamydia hijacks the ER and promotes the formation of ER-inclusion contact sites, which are important for the transfer of lipids to the inclusion47–49. During Chlamydia infection, the acetylated microtubules that surround the inclusion could enable the ER to slide towards the inclusion, thus allowing IncD/CERT to interact with the ER-resident proteins VAPA/VAPB and form ER-inclusion contact sites47,48. Although treatment with nocodazole, which disrupts microtubules, failed to prevent ER accumulation around the inclusion49, acetylated microtubules are notoriously resistant to nocodazole treatment and may still be present following treatment.
Recently, we have shown that the chlamydial protein CT813 (also called InaC50) is critical for promoting microtubule modifications during infection through its interaction with and activation of the small host GTPases ARF1 and ARF433, supporting prior data showing interactions between CT813 and ARF proteins50 (Figure 1D). It is unclear how the CT813:ARF complex is able to influence microtubule PTMs. However, inhibitors of RhoA and ROCK (Rho-associated protein kinase) decrease the number of inclusions associated with stable microtubules, suggesting that both of these proteins are involved in this process32. There is evidence demonstrating that RhoA triggers microtubule stabilization via its interaction with the mammalian homolog of Diaphanous (mDia). The activation of mDia generates capped microtubules, thus preventing catastrophic microtubule disassembly51,52. Interestingly, DIAPH2 was identified in an RNAi screen in Drosophila S1 cells as important for chlamydial inclusion development10. It would be interesting to determine whether C. trachomatis co-opts mDia to stabilize microtubules and generate the post-translationally modified microtubule cages around the inclusion, particularly since mDia is also involved in actin polymerization53.
Chlamydia trachomatis builds actin scaffolds around its inclusion to promote inclusion stability
As the inclusion continues to grow, actin and intermediate filaments associate with the inclusion54 (Figure 1E). This association increases progressively from ~20 hours PI until the end of Chlamydia’s intracellular life cycle. Disruption of the actin cytoskeleton results in the rupture of the inclusion membrane and the leakage of C. trachomatis into the host cytoplasm, demonstrating that the maintenance of the inclusion’s integrity requires intact actin cages54. Interestingly, RhoA—but not ROCK—also plays a major role in this event, as it is recruited to the inclusion and its depletion results in a substantial loss of actin scaffolds around the inclusion54.
The chlamydial protein CT813 is the only effector identified to date to regulate actin recruitment around the inclusion33,50 (Figure 1E). Interestingly, this function of CT813 appears to be independent of its role in regulating post-translationally modified microtubule cages, as the overexpression of CT813 in wild-type Chlamydia results in the loss of post-translationally modified microtubules but not actin cages33. Together, these data suggest that both CT813 and RhoA participate in actin cage formation and microtubule stabilization. While microtubule stabilization also depends on ARF1/ARF4 and ROCK, actin polymerization does not require ROCK. The role of ARF in the formation of actin cages remains unclear, as depletion of ARF does not affect actin polymerization33.
Recently, the actin cytoskeleton has been implicated in Golgi reorganization during infection. Using chemical mutagenesis, it has been suggested that CT813 organizes Golgi mini-stacks around the inclusion through the formation of actin cages50. However, a CT813-overexpressing Chlamydia strain that has actin but no post-translationally modified microtubule cages displays a defect in Golgi organization around the inclusion. This suggests that it is the CT813-dependent induction of post-translationally modified microtubule cages that controls Golgi organization33. Therefore, the exact role of the actin cytoskeleton in organelle repositioning during Chlamydia infection is still unclear.
Chlamydia uses the actin cytoskeleton to exit host cells by extrusion
Chlamydia exits the host cell through two mutually exclusive mechanisms: extrusion and cell lysis (Figure 1F, left and right, respectively). Note that to exit their host cells using the lytic process, Chlamydia must extricate themselves from the cytoskeleton structures that encase the inclusion, in particular the actin scaffold55. Pgp4, a transcription factor encoded by the chlamydial plasmid, is essential for actin depolymerization prior to cell exit, as the deletion of this regulatory gene prevents actin disassembly and completely blocks Chlamydia exit55. The chlamydial protease activity factor, which cleaves intermediate filaments, is also involved in cell exit, but it does not play a role in actin disassembly54–56.
While the lytic pathway requires actin depolymerization to proceed, extrusion is regulated by an acto-myosin-mediated mechanism57,58. Extrusion involves the protrusion of the intact membrane-bound inclusion out of the cell and the pinching off of the inclusion into a separate compartment. The resulting extruded inclusion is surrounded by the actin cytoskeleton59, the host plasma membrane, and a thin layer of cytoplasm between plasma and inclusion membranes57. Extrusion requires N-WASP-mediated actin polymerization and myosin II-dependent contraction of stable actin filaments57,58. The septin family of cytoskeletal proteins regulates actin fiber formation on the inclusion membrane60 through an unknown mechanism. Interestingly, RhoA is also involved in this process, where it specifically regulates the final stage of extrusion—pinching off and separation of the extrusion from the host cell57.
The signals that dictate whether Chlamydia exits the host cell by lysis or extrusion are not well understood. However, the Chlamydial inclusion protein CT228 has been shown to play a central role in this process58. CT228 recruits the MYPTI subunit of myosin phosphatase to microdomains on the inclusion membrane early during infection. MYPTI-mediated phosphorylation of myosin light chain II (MLC2) favors extrusion-mediated exit, while the depletion or dephosphorylation of MLC2 shifts the balance towards the lytic pathway. Thus, Chlamydia establishes local cytoskeletal signaling networks on the inclusion membrane to direct its escape from the host cell.
Concluding remarks
Chlamydia has evolved efficient mechanisms to hijack essential components of the host cytoskeleton. The successful establishment of Chlamydia’s intracellular niche and dissemination of infectious progeny relies on the proper spatial and temporal control of both actin and microtubules. To orchestrate this balance, Chlamydia employs effector proteins to recruit host proteins to the inclusion membrane and modulate the activity of host cytoskeletal signaling networks. Recent work has only begun to shed light on the identity of these chlamydial effector proteins.
Almost a decade ago, an RNAi screen in C. trachomatis-infected Drosophila cells revealed the importance of numerous host cytoskeleton-associated proteins in inclusion development, supporting the critical role of the cytoskeleton during infection10. However, limited mechanistic information regarding the role of chlamydial effector proteins in this process was available owing to the intractability of Chlamydia to genetic manipulation. Recent advances in Chlamydia genetics and the expansion of the Chlamydia genetic toolbox now provide the tools necessary to dissect the molecular pathways and the chlamydial effectors that control the interactions between the chlamydial inclusion and the host cytoskeleton. Identifying these mechanisms is important not only for understanding Chlamydia pathogenesis and developing novel therapeutics but also because it has the potential to identify new host cellular pathways that regulate the cytoskeleton.
Competing interests
The authors declare that they have no competing interests.
Grant information
This work was supported by National Institutes of Health grant AI116983 to Fabienne Paumet.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgements
We thank the reviewers for their insightful comments and criticisms of the manuscript. We apologize for the numerous scientific contributions that were not cited due to space constraints.
Faculty Opinions recommended
References
1.
WHO: World Health Organization: Global prevalence and incidence of selected curable sexually transmitted infections overview and estimates. Geneva, WHO. 2005.
2.
Gerbase AC, Rowley JT, Mertens TE:
Global epidemiology of sexually transmitted diseases.
Lancet.
1998; 351 Suppl 3: 2–4. PubMed Abstract
| Publisher Full Text
3.
CDC: Sexually Transmitted Disease Surveillance 2011. Atlanta: US Department of Health and Human Services. 2012: 1–184. Reference Source
4.
Romano Carratelli C, Nuzzo I, Cozzolino D, et al.:
Relationship between Chlamydia pneumoniae infection, inflammatory markers, and coronary heart diseases.
Int Immunopharmacol.
2006; 6(5): 848–53. PubMed Abstract
| Publisher Full Text
5.
Ezzahiri R, Stassen FR, Kurvers HR, et al.:
Chlamydia pneumoniae infections augment atherosclerotic lesion formation: a role for serum amyloid P.
APMIS.
2006; 114(2): 117–26. PubMed Abstract
| Publisher Full Text
6.
Abdelrahman YM, Belland RJ:
The chlamydial developmental cycle.
FEMS Microbiol Rev.
2005; 29(5): 949–59. PubMed Abstract
| Publisher Full Text
7.
Scidmore MA:
Recent advances in Chlamydia subversion of host cytoskeletal and membrane trafficking pathways.
Microbes Infect.
2011; 13(6): 527–35. PubMed Abstract
| Publisher Full Text
| Free Full Text
8.
Su H, Raymond L, Rockey DD, et al.:
A recombinant Chlamydia trachomatis major outer membrane protein binds to heparan sulfate receptors on epithelial cells.
Proc Natl Acad Sci U S A.
1996; 93(20): 11143–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
9.
Carabeo RA, Grieshaber SS, Fischer E, et al.:
Chlamydia trachomatis induces remodeling of the actin cytoskeleton during attachment and entry into HeLa cells.
Infect Immun.
2002; 70(7): 3793–803. PubMed Abstract
| Publisher Full Text
| Free Full Text
10.
Elwell CA, Ceesay A, Kim JH, et al.:
RNA interference screen identifies Abl kinase and PDGFR signaling in Chlamydia trachomatis entry.
PLoS Pathog.
2008; 4(3): e1000021. PubMed Abstract
| Publisher Full Text
| Free Full Text
13.
Jewett TJ, Miller NJ, Dooley CA, et al.:
The conserved Tarp actin binding domain is important for chlamydial invasion.
PLoS Pathog.
2010; 6(7): e1000997. PubMed Abstract
| Publisher Full Text
| Free Full Text
15.
Jiwani S, Alvarado S, Ohr RJ, et al.:
Chlamydia trachomatis Tarp harbors distinct G and F actin binding domains that bundle actin filaments.
J Bacteriol.
2013; 195(4): 708–16. PubMed Abstract
| Publisher Full Text
| Free Full Text
16.
Carabeo RA, Grieshaber SS, Hasenkrug A, et al.:
Requirement for the Rac GTPase in Chlamydia trachomatis invasion of non-phagocytic cells.
Traffic.
2004; 5(6): 418–25. PubMed Abstract
| Publisher Full Text
17.
Subtil A, Wyplosz B, Balañá ME, et al.:
Analysis of Chlamydia caviae entry sites and involvement of Cdc42 and Rac activity.
J Cell Sci.
2004; 117(Pt 17): 3923–33. PubMed Abstract
| Publisher Full Text
19.
Suzuki T, Mimuro H, Miki H, et al.:
Rho family GTPase Cdc42 is essential for the actin-based motility of Shigella in mammalian cells.
J Exp Med.
2000; 191(11): 1905–20. PubMed Abstract
| Publisher Full Text
| Free Full Text
22.
Jiwani S, Ohr RJ, Fischer ER, et al.:
Chlamydia trachomatis Tarp cooperates with the Arp2/3 complex to increase the rate of actin polymerization.
Biochem Biophys Res Commun.
2012; 420(4): 816–21. PubMed Abstract
| Publisher Full Text
| Free Full Text
23.
Grieshaber SS, Grieshaber NA, Hackstadt T:
Chlamydia trachomatis uses host cell dynein to traffic to the microtubule-organizing center in a p50 dynamitin-independent process.
J Cell Sci.
2003; 116(Pt 18): 3793–802. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
27.
Richards TS, Knowlton AE, Grieshaber SS:
Chlamydia trachomatis homotypic inclusion fusion is promoted by host microtubule trafficking.
BMC Microbiol.
2013; 13: 185. PubMed Abstract
| Publisher Full Text
| Free Full Text
28.
Pannekoek Y, Spaargaren J, Langerak AA, et al.:
Interrelationship between polymorphisms of incA, fusogenic properties of Chlamydia trachomatis strains, and clinical manifestations in patients in The Netherlands.
J Clin Microbiol.
2005; 43(5): 2441–3. PubMed Abstract
| Publisher Full Text
| Free Full Text
29.
Pannekoek Y, van der Ende A, Eijk PP, et al.:
Normal IncA expression and fusogenicity of inclusions in Chlamydia trachomatis isolates with the incA I47T mutation.
Infect Immun.
2001; 69(7): 4654–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
30.
Suchland RJ, Rockey DD, Bannantine JP, et al.:
Isolates of Chlamydia trachomatis that occupy nonfusogenic inclusions lack IncA, a protein localized to the inclusion membrane.
Infect Immun.
2000; 68(1): 360–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
32.
Al-Zeer MA, Al-Younes HM, Kerr M, et al.:
Chlamydia trachomatis remodels stable microtubules to coordinate Golgi stack recruitment to the chlamydial inclusion surface.
Mol Microbiol.
2014; 94(6): 1285–97. PubMed Abstract
| Publisher Full Text
33.
Wesolowski J, Weber MM, Nawrotek A, et al.:
Chlamydia Hijacks ARF GTPases To Coordinate Microtubule Posttranslational Modifications and Golgi Complex Positioning.
mBio.
2017; 8(3): pii: e02280-16 PubMed Abstract
| Publisher Full Text
| Free Full Text
35.
Webster DR, Gundersen GG, Bulinski JC, et al.:
Differential turnover of tyrosinated and detyrosinated microtubules.
Proc Natl Acad Sci U S A.
1987; 84(24): 9040–4. PubMed Abstract
| Publisher Full Text
| Free Full Text
37.
Carabeo RA, Mead DJ, Hackstadt T:
Golgi-dependent transport of cholesterol to the Chlamydia trachomatis inclusion.
Proc Natl Acad Sci U S A.
2003; 100(11): 6771–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
38.
Hackstadt T, Rockey DD, Heinzen RA, et al.:
Chlamydia trachomatis interrupts an exocytic pathway to acquire endogenously synthesized sphingomyelin in transit from the Golgi apparatus to the plasma membrane.
EMBO J.
1996; 15(5): 964–77. PubMed Abstract
| Free Full Text
39.
Kumar Y, Cocchiaro J, Valdivia RH:
The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets.
Curr Biol.
2006; 16(16): 1646–51. PubMed Abstract
| Publisher Full Text
40.
Beatty WL:
Trafficking from CD63-positive late endocytic multivesicular bodies is essential for intracellular development of Chlamydia trachomatis.
J Cell Sci.
2006; 119(Pt 2): 350–9. PubMed Abstract
| Publisher Full Text
42.
Lin SX, Gundersen GG, Maxfield FR:
Export from pericentriolar endocytic recycling compartment to cell surface depends on stable, detyrosinated (glu) microtubules and kinesin.
Mol Biol Cell.
2002; 13(1): 96–109. PubMed Abstract
| Publisher Full Text
| Free Full Text
45.
Gambarte Tudela J, Capmany A, Romao M, et al.:
The late endocytic Rab39a GTPase regulates the interaction between multivesicular bodies and chlamydial inclusions.
J Cell Sci.
2015; 128(16): 3068–81. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
46.
Friedman JR, Webster BM, Mastronarde DN, et al.:
ER sliding dynamics and ER-mitochondrial contacts occur on acetylated microtubules.
J Cell Biol.
2010; 190(3): 363–75. PubMed Abstract
| Publisher Full Text
| Free Full Text
49.
Dumoux M, Clare DK, Saibil HR, et al.:
Chlamydiae assemble a pathogen synapse to hijack the host endoplasmic reticulum.
Traffic.
2012; 13(12): 1612–27. PubMed Abstract
| Publisher Full Text
| Free Full Text
51.
Palazzo AF, Cook TA, Alberts AS, et al.:
mDia mediates Rho-regulated formation and orientation of stable microtubules.
Nat Cell Biol.
2001; 3(8): 723–9. PubMed Abstract
| Publisher Full Text
53.
Copeland JW, Treisman R:
The diaphanous-related formin mDia1 controls serum response factor activity through its effects on actin polymerization.
Mol Biol Cell.
2002; 13(11): 4088–99. PubMed Abstract
| Publisher Full Text
| Free Full Text
54.
Kumar Y, Valdivia RH:
Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds.
Cell Host Microbe.
2008; 4(2): 159–69. PubMed Abstract
| Publisher Full Text
| Free Full Text
56.
Snavely EA, Kokes M, Dunn JD, et al.:
Reassessing the role of the secreted protease CPAF in Chlamydia trachomatis infection through genetic approaches.
Pathog Dis.
2014; 71(3): 336–51. PubMed Abstract
| Publisher Full Text
| Free Full Text
58.
Lutter EI, Barger AC, Nair V, et al.:
Chlamydia trachomatis inclusion membrane protein CT228 recruits elements of the myosin phosphatase pathway to regulate release mechanisms.
Cell Rep.
2013; 3(6): 1921–31. PubMed Abstract
| Publisher Full Text
| Free Full Text
59.
Chin E, Kirker K, Zuck M, et al.:
Actin recruitment to the Chlamydia inclusion is spatiotemporally regulated by a mechanism that requires host and bacterial factors.
PLoS One.
2012; 7(10): e46949. PubMed Abstract
| Publisher Full Text
| Free Full Text
60.
Volceanov L, Herbst K, Biniossek M, et al.:
Septins arrange F-actin-containing fibers on the Chlamydia trachomatis inclusion and are required for normal release of the inclusion by extrusion.
mBio.
2014; 5(5): e01802–14. PubMed Abstract
| Publisher Full Text
| Free Full Text
This work was supported by National Institutes of Health grant AI116983 to Fabienne Paumet
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Wesolowski J and Paumet F. Taking control: reorganization of the host cytoskeleton by Chlamydia [version 1; peer review: 5 approved]. F1000Research 2017, 6(F1000 Faculty Rev):2058 (https://doi.org/10.12688/f1000research.12316.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Meyer T and Al-Zeer M. Reviewer Report For: Taking control: reorganization of the host cytoskeleton by Chlamydia [version 1; peer review: 5 approved]. F1000Research 2017, 6(F1000 Faculty Rev):2058 (https://doi.org/10.5256/f1000research.13333.r26845)
We confirm that we have read this submission and believe that we have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
We confirm that we have read this submission and believe that we have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Meyer T and Al-Zeer M. Reviewer Report For: Taking control: reorganization of the host cytoskeleton by Chlamydia [version 1; peer review: 5 approved]. F1000Research 2017, 6(F1000 Faculty Rev):2058 (https://doi.org/10.5256/f1000research.13333.r26845)
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)