Keywords
multiple system atrophy, MSA, neurodegenerative disorders, α-synuclein
multiple system atrophy, MSA, neurodegenerative disorders, α-synuclein
Multiple system atrophy (MSA) is a neurodegenerative movement disorder affecting around 1 in 20,000 people1,2. It occurs sporadically, usually presenting between the age of 35 and 65 years with a variable combination of parkinsonian, cerebellar, and autonomic features and rapidly progressing for 9 years on average3–6. According to the presenting clinical features and predominant manifestations, MSA is usually categorised as MSA-C or MSA-P, there can be mixed signs, and some patients present with autonomic features. Familial MSA has been reported but in only a handful of cases worldwide. Research toward potential treatments for MSA, as with many rare diseases, has been limited, resulting in a paucity of knowledge regarding its underlying causes. Initial clues came from studying α-synuclein (SNCA) and the hallmark histopathology in the brains of patients with MSA: glial cytoplasmic inclusions (GCIs) that reside predominantly in oligodendrocytes, the post mortem identification of which is required for a definitive diagnosis. Besides MSA, the only conditions that have GCIs in the brain are certain families with SNCA mutations. Three groups found that the GCIs contain abnormal forms of SNCA protein7–12, the same protein that accumulates in Parkinson’s disease (PD) and dementia with Lewy bodies12. These studies were motivated by a link between point mutations in the SNCA gene and heritable forms of PD13,14. The similarities between MSA and PD have proven more complicated to disentangle, as SNCA mutations in some families clinically and pathologically resemble MSA and others even have features of frontal dementia with severe pathology15.
It has been nearly two decades since MSA was characterised as a synucleinopathy, and apart from the PD-MSA overlap identified in SNCA families, researchers have not been able to further understand the aetiology of MSA or alter or halt the disease process. This brief review will set out the progress that has been made in recent years toward understanding the pathomechanisms of SNCA aggregation and toxicity in relation to MSA, particularly the emerging hypotheses of aetiology based on genetic studies. As clinical trials targeting SNCA proceed in PD and MSA, there is increasing urgency to better understand its relevant cellular interactions in parallel with the development of sensitive biomarkers capable of diagnosing patients at an earlier disease stage.
Despite the key involvement of abnormal SNCA processing, misfolding, and aggregation in synucleinopathies, the normal function of the protein is not fully understood. It is a peripheral membrane protein, localized at nerve terminals where it is thought to play a role in the release of neurotransmitters, and recently has been reported to enhance transient synaptic vesicle fusion16,17 and possibly disrupt the support and maintenance of neurons provided by oligodendrocytes. In MSA, SNCA is deposited widely, but there are more severely affected regions such as the basal ganglia, cerebellum, pons, inferior olivary nuclei, and spinal cord18,19. Not only is SNCA deposition clearly distinguishable between MSA and PD cases but MSA-like pathology underlies both cerebellar (MSA-C) and parkinsonian (MSA-P) manifestations. There is also minimal change MSA pathology in some cases that have a longer disease duration. How this single protein can apparently be the culprit in these different disease phenotypes, with such varied localization in different cell types and brain regions, is an unresolved question.
One compelling explanation for the clinicopathological diversity in the synucleinopathies is that distinct strains of SNCA are responsible for generating heterogeneity20. These conformational variants include different oligomer combinations, fibrils and ribbons, although their relative contribution to the anatomical distribution and deposition of SNCA in MSA and other synucleinopathies and the formation of GCIs in MSA has yet to be determined. Furthermore, it has been posited that the specific structure of SNCA derived from inclusions in the brains of patients with MSA is especially toxic, capable of propagating to adjacent cells and inducing neurodegeneration when injected into transgenic mice, akin to the permissive templating of prion protein and even prompting reclassification of MSA as a prion disease21,22. However, it remains to be shown conclusively that oligodendroglial MSA-type pathology is provoked by seeded aggregation of SNCA.
The pathomechanisms of MSA are being steadily elucidated as studies examine the molecular interactions of SNCA with other proteins in MSA. A recent study23 has reported that SNCA engages with proteins that regulate autophagy in the MSA brain, implicating cellular degradation as central to the pathogenesis of MSA and potentially unifying it with other neurodegenerative diseases for the purpose of therapeutic intervention of these pathways23,24. Additionally, there is an emerging conviction that SNCA induces deficits in myelination25 and there is a possible role for inflammatory/apoptotic mechanisms.
The initial genome-wide association study (GWAS) in PD yielded significant association at the SNCA and microtubule-associated protein tau (MAPT) genes26. Common variation in the gene encoding SNCA was first identified as a risk factor for MSA in 200927, but the association of variants across SNCA in different populations was not replicated in later studies28–32 and was thought to be due to a mixed control population used in the initial studies. The first GWAS to be conducted in MSA yielded negative results around the SNCA locus28. As mentioned earlier, several SNCA point mutations14,33–39 and SNCA gene duplications40, triplications41,42, and double duplications43 have been associated with familial forms of PD (Figure 1 and Table 1 and Table 2). Some of these families have manifestations of both PD and MSA and have clinical signs or neuropathological features or both44,45. In particular, the A53T, A53E, and G51D mutations and SNCA gene triplications are associated with a more aggressive MSA-like clinical and pathological phenotype45 (See Table 1 and Table 2 for details of the clinical and neuropathological features of SNCA mutations). Exactly why the codon 51 and 53 mutations in the SNCA gene lead to an MSA-like clinical and pathological phenotype is not known, but this is likely to be associated with the importance of this defined region and toxic gain of function of these protein changes (Figure 1)46. From a clinical perspective, if there is any hint of a family history in patients with MSA, then the SNCA gene should be sequenced by using traditional Sanger47, gene panel, or exome sequencing and analysed for copy number changes48.
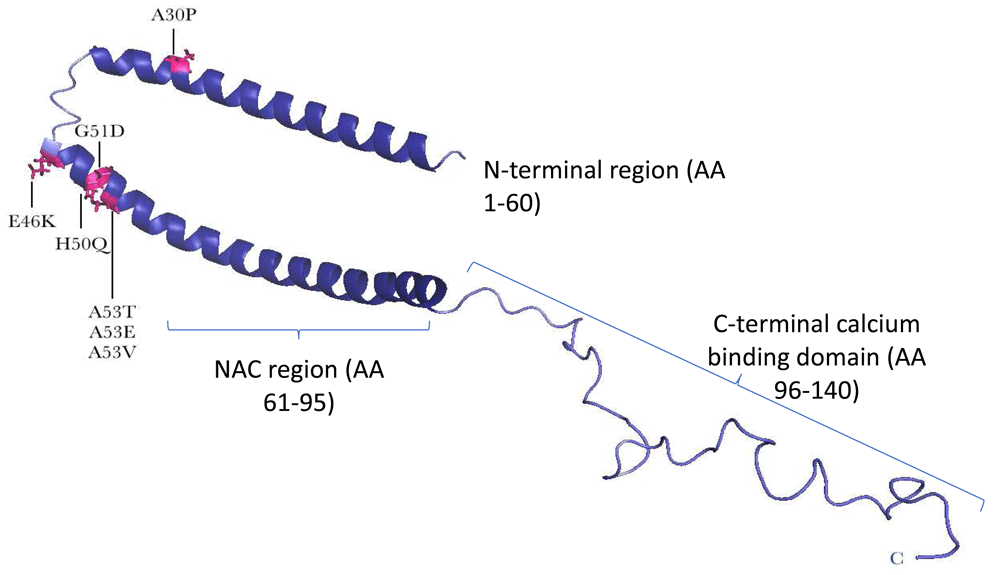
A structure of the full-length, membrane-bound form of alpha-synuclein (SNCA) protein reveals a conformation in which the N-terminal two-thirds of the protein forms a broken, amphipathic alpha-helix. This structured portion of the protein is responsible for membrane binding, and residues at the very N-terminus are essential for this process. In the nuclear magnetic resonance structure of SNCA, the negatively charged C-terminal tail remains flexible and disordered (based on Yu et al.46). The positions of point mutations associated with Parkinson’s disease are indicated with arrows and in pink. All mutations are heterozygous, except for p.A53V, which is homozygous.
| SNCA protein change | p.Ala30Pro1 | p.Glu46Lys2 | p.His50Gln3 | p.Gly51Asp4,5 | p.Ala53Thr6 | p.Ala53Glu7 | p.Ala53Val8 | Duplication9 | Triplication10 | Double duplication11 |
|---|---|---|---|---|---|---|---|---|---|---|
| SNCA: 5′ start - ATG start - | 177G>C c.88G>C | 225G>A c.136G>A | 239T>G c.150T>G12 | 241G>A c.152 G>A | 246G>A c.157G>A | 247C>A c.158C>A | 247C>T c.158C>T | Whole gene copy number | Whole gene copy number | Whole gene copy number |
| Zygosity | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Homozygous/ heterozygous | Gene duplication on one allele | Gene triplication on one allele | Gene duplication on both alleles |
| Clinical phenotype | Classic PD | Dementia with Lewy bodies phenotype | Classic PD | Severe PD with some patients with MSA features | Severe PD with some patients with MSA features | Severe PD with some patients with MSA features | Homozygous = PD Heterozygous = cognitive decline or psychosis | Usually classic PD, some with severe cognitive and frontal dementia13 | Severe PD with some patients with MSA features | Severe PD |
| Family | German14 | Spanish Basque Country15 | English16 | British4, French17, and Japanese18 | Large Sicilian (Contursi) kindred6 and Greek19, Swedish20, Korean21 | Finnish22 | Japanese23 | French24, Italian25, Japanese26,27, Korean28, Swedish29, UK, Welsh13 | Spellman- Muenter (Iowa) kindred30, Swedish9 | Pakistani31 |
| Estimated penetrance/ risk | 71.4% | 30% | 30% Heterozygous ExAc =4/121,306 | 100% | 85% | 100% | Homozygous 100% Heterozygous on ExAc = 1/121,304 | 44%27 | 100% | 100% |
| Mean age of onset, years | 60 | 50–65 | 71 | 44 (as early as 19)5 | 48 | 43 | 59 | 50 | 40 | 31 |
| Clinical symptoms | Progressive parkinsonism, walking difficulties, no other non-motor symptoms except cognitive decline (50% of patients) | Resting tremor, bradykinesia, postural instability, severe immobility, dementia, and visual hallucinations | Resting hand tremor, benign course | Resting tremor, dystonia, cognitive/frontal decline, anxiety, depression, visual hallucination, and autonomic disturbances5 | Moderate tremor, rigidity, bradykinesia, postural instability, severe dementia, depression, and autonomic disturbance32 | Bradykinesia, resting tremor, rigidity, insomnia, spasticity, myoclonic jerks, anxiety, and panic disorders | Bradykinesia, resting tremor, rigidity, mild cognitive decline, visual hallucination, sleep disorder, delusions, and paranoia | Bradykinesia, resting tremor, rigidity, mild asymmetric onset. Some have epilepsy, depression, and frontal dementia. | Severe early onset parkinsonism, resting tremor, bradykinesia, rigidity, and symptoms of MSA | Severe bilateral bradykinesia rigidity, mild resting tremor, severe depression, and postpartum psychosis |
| Sustained response to levodopa | Transient marked, developed hallucinations | Mild to moderate, developed hallucinations and conscious fluctuation | Moderate | Transient marked, developed choreiform movements | Marked | Marked, developed dyskinesia33 | Marked | Mild to moderate, developed motor fluctuation and dyskinesia | Marked | N/A |
ExAC, http://exac.broadinstitute.org; MSA, multiple system atrophy; N/A, no data available; PD, Parkinson’s disease; SNCA, alpha-synuclein gene.
| SNCA protein change | p.Ala30Pro1 | p.Glu46Lys2 | p.His50Gln3 | p.Gly51Asp4,5 | p.Ala53Thr6,34 | p.Ala53Glu7 | Duplication35 | Triplication10 |
|---|---|---|---|---|---|---|---|---|
| Neuropathology summary | Widespread synuclein neuropathology and LBs | Widespread synuclein and ubiquitin neuropathology and LBs | Widespread synuclein neuropathology and LBs | Widespread synuclein neuropathology, LBs, and GCIs | Widespread synuclein neuropathology, LBs, and GCIs | Widespread synuclein neuropathology, LBs, and GCIs | Widespread synuclein neuropathology, LBs, and some GCIs | Widespread synuclein neuropathology, LBs, and GCIs |
| LBs | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Cortical neuronal loss | Widespread | Widespread | Not identified | Widespread, moderate to severe, anterior temporal, piriform, and insular cortices4 | Widespread, moderate to severe, mid- frontal cortex, superior temporal cortex, and inferior parietal cortex | Widespread and severe | Not identified | Widespread |
| Hippocampal neuronal loss | CA1/2/3 | Not identified | Not identified | Severe in CA2/34 | Severe in CA1, moderate in CA2/334 | Mild in CA1/2/3 | CA2/3 region | CA2/3 region |
| Brain stem neuronal loss | Substantia nigra, locus coeruleus, and dorsal nuclei of the vagus | Substantia nigra, locus coeruleus, and dorsal nuclei of the vagus | Substantia nigra | Substantia nigra, locus coeruleus, and dorsal motor nuclei of vagus | Substantia nigra and locus coeruleus | Substantia nigra | Substantia nigra, locus coeruleus, and dorsal motor nuclei of vagus | Substantia nigra and locus coeruleus |
| Neuronal α-synuclein pathology | Globular, spherical, reniform, widespread om cortex and brainstem | Concentric, nonconcentric, granular | PD-type (Braak stage 6) | Annular, crescentic, globular, diffuse, NFT-like, Widespread cortical | Wide spread cortex and brain stem, LBs | Annular, annular, LB-like inclusions, mild in brainstem | PD-type (Braak stage 6), widespread from brainstem and neocortex | Widespread in cortex, few in brainstem |
| Glial α-synuclein pathology | No | No | No | GCI-like | GCI-like | Granular GCI | GCI-like | Atypical LBs, GCIs |
| Phosphorylated tau Braak and Braak stage | II | Not identified | III | IIa | I | N/A | I | N/A |
| Aβ deposition | Thal phase 136 | Neocortical | Neocortical | N/A | N/A | N/A | Sparse neocortical | N/A |
A number of PD risk factors have not been replicated in MSA4,6,48–61, but other disorders such as spinocerebellar ataxia type 17 and progressive supranuclear palsy62–64 can mimic MSA in the early stages and should be included in clinical and genetic testing. In a statistical analysis of 5,302 patients with PD and 4,161 controls from 15 sites, Elbaz and colleagues found no evidence for an interactive effect between the H1 haplotype in the MAPT gene and single-nucleotide polymorphisms in the SNCA gene on disease65. Variation in each gene was associated with PD risk, indicating independent effects. In MSA, the H1 haplotype has been associated with MSA66 and the MAPT gene was also implicated in the MSA GWAS28. Familial inheritance of MSA is rare but has been observed. These families often have atypical clinical features, and the genetic analysis led to the discovery of mutations in the COQ2 gene, which plays a role in synthesising the mitochondrial electron transporter and antioxidant coenzyme Q10. These mutations were posited to impair the activity of the mitochondrial respiratory chain and increase oxidative stress, implicating COQ2 variants as a risk factor for sporadic MSA58. Though initially promising, these findings have not been consistently replicated in various populations, refuting COQ2 polymorphisms as common MSA risk factors58. Nonetheless, this has turned attention, and emerging hypotheses centre on mitochondrial dysfunction as a central component of the pathophysiological cascade in MSA67.
The first GWAS in MSA was carried out by Sailer and colleagues and was extremely important but challenging given the rarity of MSA28. At just under 1,000 MSA cases, the analysis was still statistically under-powered28. Studies that are more highly powered are needed to follow up on the importance of the three genes identified that were flagged for being associated: FBXO47, ELOVL7, and MAPT28. It will be important to follow this GWAS up with greater numbers of MSA cases, analyse age at onset association68–70, and employ advanced transcriptome sequencing in MSA patient brain tissue to assess the associated genes and other genes thought to be involved in MSA, such as immune-responsive and iron metabolism genes71,72.
Accurate and early diagnosis of MSA continues to be an important research objective as the heterogeneous features of PD and other atypical parkinsonism syndromes can mimic MSA. One retrospective clinicopathological study revealed that 38% of patients were misdiagnosed with MSA on the basis of expert interpretation of their symptomatic presentation73–75. Genetic analysis will be important to identify the rare MSA cases with SNCA mutations and to help differentiate MSA from similar disorders such as spinocerebellar ataxia type 1762,64. Thus, biomarkers that are more sensitive are imperative to improve diagnosis and enlist individuals with the appropriate disease in clinical trials. This will be imperative in the development of effective treatments for the MSA patient population. Both α-synuclein and CoQ10 are being pursued as potential therapeutic targets, and international collaborative study groups are promoting this work with CoQ10 supplementation, the preparation of α-synuclein antisense oligonucleotide, and immunisation trials to be conducted in PD and MSA patients by either intravenous or intrathecal routes.
Until disease-modifying treatments become available, symptom management will remain the mainstay of care for patients with MSA. Patient support organisations such as the MSA Trust (www.msatrust.org.uk/) and the MSA coalition (https://www.multiplesystematrophy.org/) and their clinical nurse specialists are essential in providing support and advice on patient care in this rare disorder. The established drugs for controlling parkinsonism, such as L-dopa, can be effective in the early stages of MSA but often worsen the symptoms due to hypotension later in the disease. A rational treatment, based on the pathophysiology of MSA and perhaps repurposed from PD trials, needs to be developed to offer patients with MSA hope for this devastating disorder.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)