Keywords
Chikungunya virus, Public health, sequences, information system, phylogenetic analysis.
This article is included in the Neglected Tropical Diseases collection.
Chikungunya virus, Public health, sequences, information system, phylogenetic analysis.
Chikungunya virus (CHIKV) is an arbovirus (arthropod-borne virus), which is part of the Alphavirus genus and belongs to the Togaviridae family. It is vectored by Aedes mosquitoes and infects humans in tropical and sub-tropical areas of Asia and Africa1. CHIKV has a positive-sense, single-stranded RNA genome of 12 kb, which can persist for years in humans. Symptoms include rash and febrile illness associated with severe arthralgia2.
Currently, the following geographical genotypes have been identified through phylogenetic analysis of CHIKV E1 gene sequences: the West African (WAf), East/Central/South African (ECSA), and Asian genotypes3–5. However, most CHIKV phylogenetic studies have just used fragmented sequences from the glycoprotein envelope of the E1 gene, which avoids accurate assessments regarding the relations between strains and their evolutionary dynamics.
Recently, some complete sequences from the CHIKV genome have been made available, so we have used the available data for a complete E1 gene to develop an automated computational algorithm that can be used for accurate and rapid identification of this pathogen.
The online software tool that we have created, called Pathogen Sequence Signature Analysis (PSSA), will allow researchers to analyze nucleotide and amino acid sequence variations between sample CHIKV sequences taken from infected patients, and determine the corresponding genotype from phylogenetic analysis of the results. PSSA also provides various ways to visualize the data in order to aid understanding and interpretation of results.
To build PSSA, we used standard computer-based tools, programming languages, and infrastructure systems. PSSA is based on the Object Oriented Paradigm; thus, for its design, we used the Unified Modelling Language (UML)6. For its development, we used version 7.1.0 of the PHP language (https://www.php.net/) supported by the application server Apache version 2.4.23 (http://www.apache.org/). PSSA’s front end was developed based on version 3.3.7 of Bootstrap (http://getbootstrap.com/). Using Bootstrap is very convenient for this project because it is a framework that properly integrates JavaScript, CSS, and HTML for creating responsive web applications. PSSA uses the version 3.1.1 of the library JQuery (https://www.jquery.com/) for facilitating the use of JavaScript functionalities. After PSSA performs an analysis, it provides results in several formats. One result corresponds to an automatically generated report in pdf format, created based on version 0.0.8 of the PHP library ezpdf (https://github.com/rebuy-de/ezpdf). The other results are a force-directed graph, a radial tree, and a cartesian tree, which were developed supported by version 3.5.17 of the online JavaScript library called Data-Driven Documents, known as D3 (https://www.d3js.org/). D3 provides services for deploying data via interactive visualizations.
The geographical genotype of a sample sequence was determined based on well-defined phylogenetic clusters whose origins have been linked to a given geographic region. We analyzed all available whole genomes in GenBank database (www.ncbi.nlm.nih.gov/genbank/). However, since the E1 gene has been previously used in several studies, including Nunes et al.7, Laiton-Donat et al.8, and Volk et al.9, to determine the genotype of sample sequences, we decided to use the E1 gene. In addition, we performed extensive testing to be sure that our reference strains can accurately classify other sequences.
PSSA stores all information related to nucleotide and amino acid analysis in one relational database. In this project we created said database using version 5.7.16 of the MySql community server (http://dev.mysql.com/downloads/). The database was designed to handle the required information of the proposed nucleotide and amino acid analysis, as well as phylogenetic analyses. The database is managed using version 4.6.5.2 of phpMyAdmin (https://www.phpmyadmin.net/), which is a project that serves to administrate databases that use MySql. MySQL community also offers MySqlWorkbench (https://www.mysql.com/products/workbench/), of which version 6.3 was used to design PSSA’s database.
In addition, version 4.1 of the Integrated Development Environment (IDE) EclipsePHP (https://eclipse.org/pdt/) was used to develop PSSA. EclipsePHP allows for creation of PHP-based projects supporting PHP, CSS, and JavaScript languages. In addition, EclipsePHP provides git services for storing projects in desired repositories. Thus, we started the development of PSSA by creating a PHP project in EclipsePHP; next, we created all required files and wrote the source code for the algorithms that performed the desired analyses; and finally, we created a git configuration to store the project in the GitHub repository system. To host PSSA, the SUSE Linux Enterprise Server 12 SP2 (https://www.suse.com) was used. This server includes all applications mentioned above that are necessary for PSSA to operate correctly.
The various libraries, frameworks, and software we used to develop PSSA are all under the GNU General Public License (http://www.gnu.org/licenses/licenses.en.html). This means that only free software was used to develop our software tool.
As reference sequences we used ECSA genotype HM045811-Ross, Asian genotype HM045810, and West African genotype HM045807, searching through all nucleotide sequences of the CHIKV E1 gene that are available on the GenBank database (www.ncbi.nlm.nih.gov/genbank/). (the first isolated identified of three genotypes).
The accession numbers for the representative or alternative CHIKV E1 sequences used in the phylogenetic analysis are as follows:
ECSA genotype: HM045823, AM258993, EF012359, AM258991, AB455494, GU199352, FJ445426, FN295485, GU301781, HM045784, HM045822, KP164568, KP164570, KP164569.
Asian genotype: HM045813, HM045800, HM045790, HM045789, EF027140, EF027141, FN295483, L37661, EF452493, FJ807897, HE806461, KF318729, KJ451622, AB860301, KP164567, KP164572, KP164571, KJ451624, KP851709, KT211035, KT211049.
West African genotype: HM045816, HM045785, HM045815, HM045818, AY726732, HM045820, HM045817.
PSSA has been developed to be run in with Google Chrome, Mozilla Firefox, Internet Explorer, and Safari; nevertheless, PSSA might run in other browsers such as Opera. To run PSSA the URL http://pssa.itiud.org must be typed in the web browser.
However, if the user wants to run PSSA locally, the following steps need to be followed:
1. Download PSSA from the github repository: https://github.com/florezfernandez/pssa.
2. Install the local server the software: Apache, PHP, MySql, and PHPMyAdmin (optional).
3. Run the database script, which is available when the project is restored from the repository.
4. The project contains a file called “connection.php” in which the information regarding the connection to the database is configured. Update the information of the server, database, database user, and database password with the information of the local server. The default values provided with PSSA are: server = localhost, database = pssa, database user = “root”, and database password = “”.
The most important feature of PSSA is that the algorithm has been developed to analyze sequences taken from multiple patients. Several sequences can also be submitted per patient. The analysis process is carried out via the following steps:
1. Once the user has accessed PSSA by using the corresponding web address, they must access the “Sequence Analysis” menu, in which the user can select the menu item “Chikungunya Virus”. PSSA then presents the name, description, reference and alternative sequences of available gene(s) for CHIKV (Figure 1). For each gene (e.g., E1), two icons appear on the right side. The first icon deploys reference sequences while the second icon the alternative sequences.
2. There are two different types of analysis in PSSA. By selecting the reference sequences, users proceed with the mutation analysis, which analyzes nucleotide and amino acid changes in patient sequences, and by selecting alternative sequences users proceed with the phylogenetic analysis, which establishes the phylogenetic relationship between the submitted sequences and determines which genotype they belong to.
3. Once the type of analysis has been selected, the user can provide the patient sequences through FASTA files. PSSA includes an example dataset that can be used to test the system. The symbol '-' can be included in desired sequences in order to specify possible missing data; but tabs, blank spaces, and any other symbols than the ones used in these kind of sequences (i.e., A, C, T, G) are not accepted.

The first icon represents the reference sequence, while the second icon represents the alternative sequences of the CHIKV E1 gene. By selecting the reference sequences, users proceed with the mutation analysis, and by selecting alternative sequences users proceed with the phylogenetic analysis,
After patient sequences have been provided, the analysis algorithm is run and the system presents the corresponding results.
For the mutation analysis the system provides an online report that includes the nucleotide and corresponding amino acid changes in each patient’s sequence (Figure 2). The report can be sent to the user via e-mail in pdf format, and contains both a summary of the results and complete details of the analysis. It also provides a force-directed graph which presents each sequence as a node and where the set of nodes deployed using the same color represents one patient (Figure 3). In addition, nodes that belong to each patient are clustered based on the number of nucleotide and amino acid changes. When a sequence contains a substantial part of the E1 gene, these results are reliable and can be used for further purposes. Users might confirm that results are reliable by reading the pdf report and comparing it to the force-directed graph.
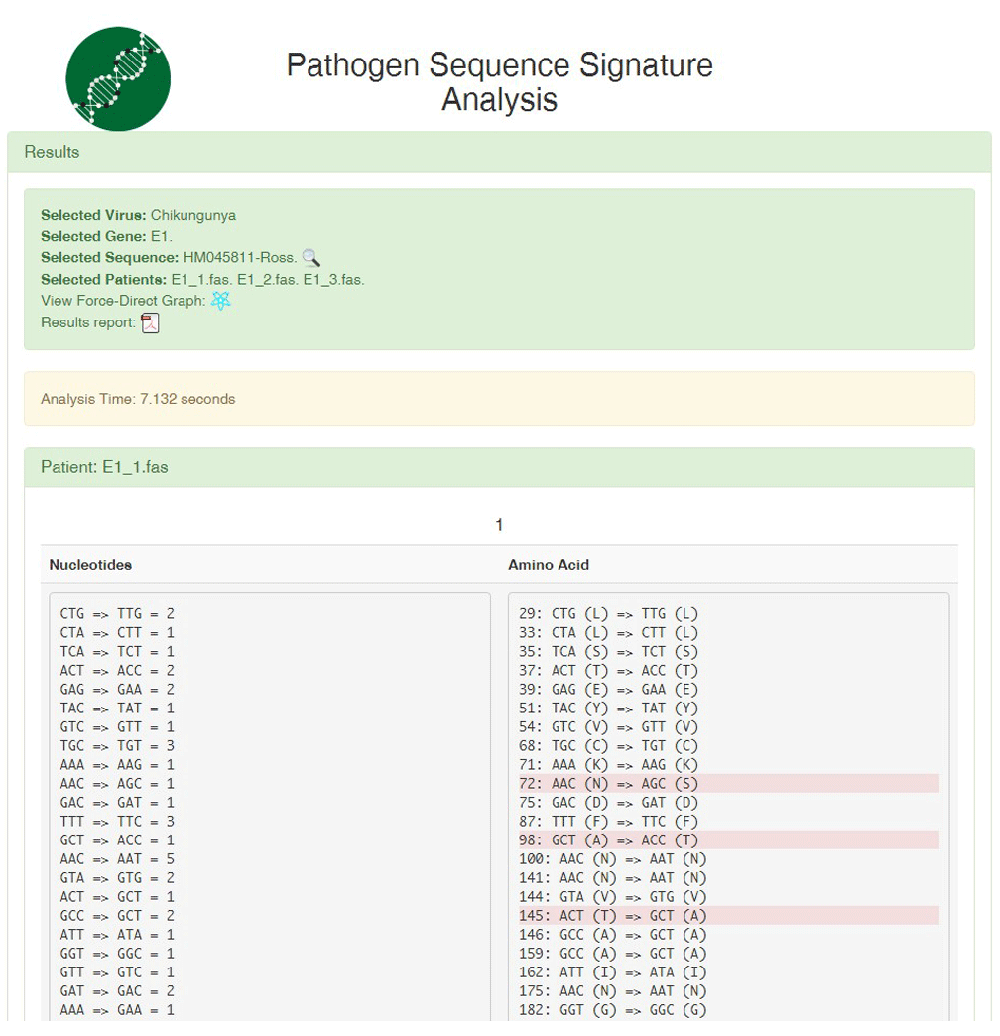

Force-directed graph presents each sequence as a node and each set of nodes of the same color as one patient. In addition, nodes that belong to each patient are clustered based on the number of nucleotide and amino acid changes.
For the phylogenetic analysis, the system presents results as a radial tree (Figure 4) and a cartesian tree (Figure 5) to establish the phylogenetic relationship between sequences and determine which genotype they belong to, based on an array of alternative sequences corresponding to the E1 gene.
The algorithm is an iterative process. Thus, for each patient file, all sequences are collected by the algorithm; then, for each sequence, some instructions of the algorithm are used to compare the iterated sequence to the selected reference sequence as well as to the alternative sequences. All information regarding the analysis is stored and used to generate the reports through the visualizations described above. It is important to mention that there are two different types of analysis in PSSA, even though they are both closely related. The mutation analysis presents results as a report of the nucleotide and amino acid variations in each patient’s sequence and a force-directed graph, whilst the phylogenetic analysis is based on the nucleotide substitution model and results are presented as a radial and cartesian tree to establish the phylogenetic relationship between sequences and determine which genotype they belong to.

The three genotypes are separated into the different branches, where each branch corresponds to one sequence obtained from the GenBank database.
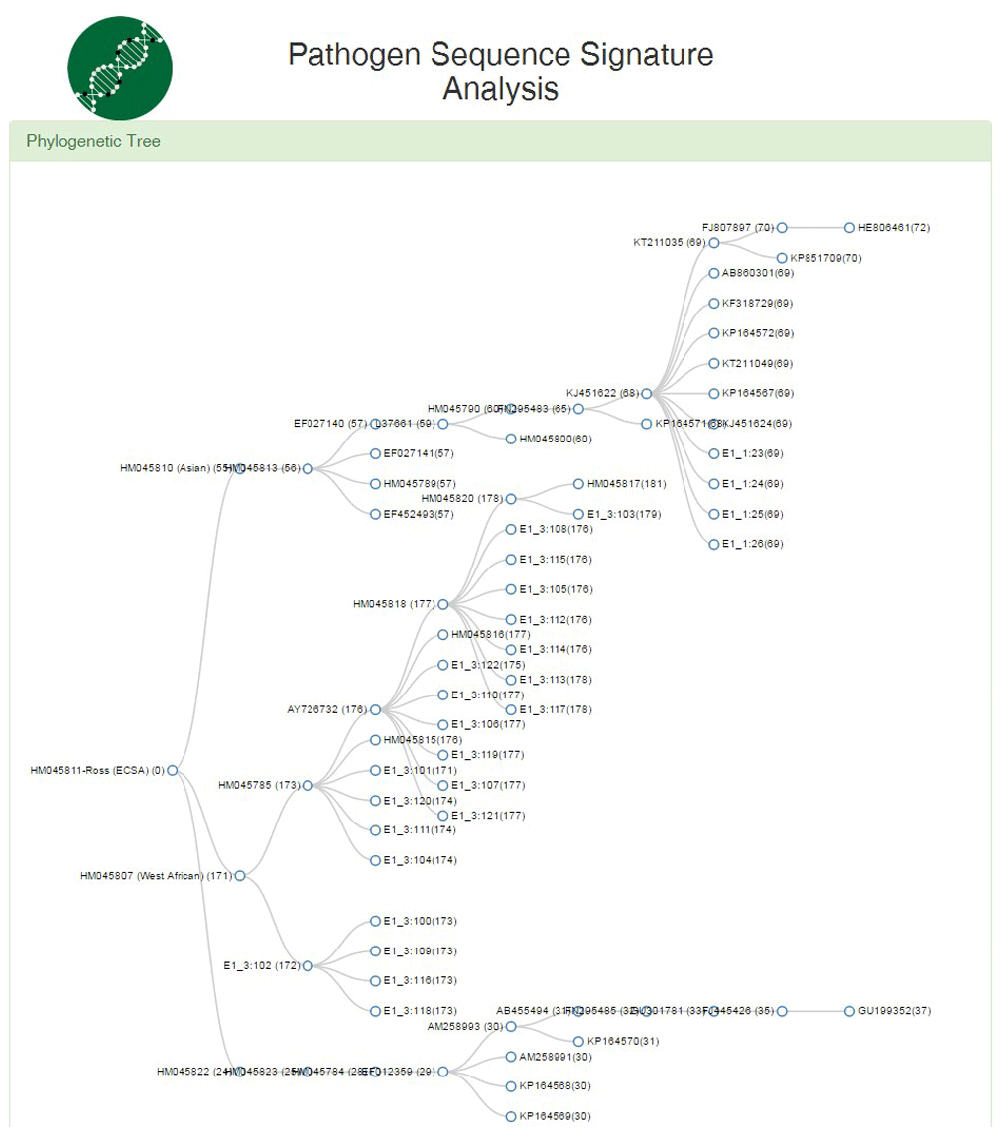
It presents the same information as the radial tree, but it shows the three genotypes separated into the different branches more clearly.
PSSA is an online software tool that provides an automated computational algorithm that guarantees accurate and reliable detection of nucleotide and amino acid sequence variations and provides various ways to visualize the data in order to aid understanding and interpretation of results.
PSSA is different to BMA10, which is another analysis tool developed in our research group, because it not only provides information regarding nucleotides and amino acid changes, but it also compares the sequences with multiple alternative sequences to identify the genotype in a phylogenetic tree.
PSSA will be useful for the identification of circulating CHIKV genotypes in an outbreak and public health surveillance. It is a flexible tool, which implies that it could be used for evaluating other microorganisms, such as bacteria (e.g., Mycobacterium tuberculosis), parasites (e.g., Leishmania) or other viruses (e.g., Dengue, Zika).
Software available from: http://pssa.itiud.org
Latest source code: https://github.com/florezfernandez/pssa
Archived source code as at the time of publication:
http://dx.doi.org/10.5281/zenodo.17992211
License: GNU General Public License (GPL)
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)