1Unit for Human Genetics, All India Institute of Speech and Hearing, Manasagangothri, India 2Department of Clinical Services, All India Institute of Speech and Hearing, Manasagangothri, India 3Department of Speech Language Pathology, All India Institute of Speech and Hearing, Manasagangothri, India
Microcephaly is a genetically heterogeneous disorder and is one of the frequently notable conditions in paediatric neuropathology which exists either as a single entity or in association with other co-morbidities. More than a single gene is implicated in true microcephaly and the list is growing with the recent advancements in sequencing technologies. Using massive parallel sequencing, we identified a novel frame shift insertion in the abnormal spindle-like microcephaly-associated protein gene in a client with true autosomal recessive primary microcephaly. Exome sequencing in the present case helped in identifying the true cause behind the disease, which helps in the premarital counselling for the sibling to avoid future recurrence of the disorder in the family.
Corresponding author:
Srinivas Kovvali
Competing interests:
No competing interests were disclosed.
Grant information:
Grants to carry out the present project were provided by All India Institute of Speech and Hearing, an autonomous Institute under Ministry of Health and Family Welfare, Government of India, India
Microcephaly with no other anomalies in the brain structure is termed as true microcephaly or autosomal recessive primary microcephaly (MCPH), where the pathology of brain is generally congenital and static with mild to moderate intellectual disability (ID) (http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=2512). Microcephaly is seen in numerous syndromes1 and even in true microcephaly, there is possibility of more than one gene implicated2,3, thus screening for a single gene may not be very fruitful in this population. Recently, whole exome sequencing (WES) has emerged as a potential approach to delineate the molecular pathology in the microcephaly population with ID1. Using WES, we report a de novo frame shift (insertion) mutation in the calponin-homology domain of ASPM (Abnormal Spindle-Like, Microcephaly-Associated) gene which is a candidate for MCPH5 (Microcephaly 5, primary, autosomal recessive) in a client with true microcephaly.
Report
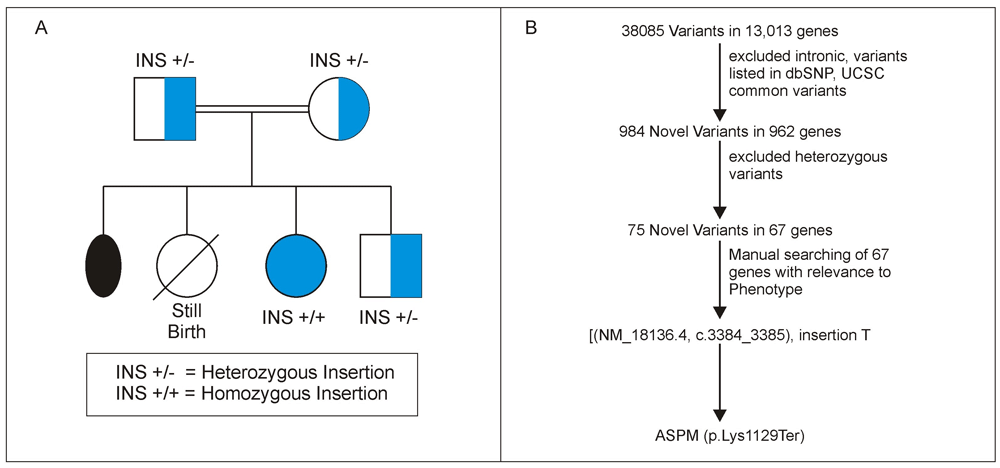
The client sequenced was under the research project cerebral palsy and spectrum conditions (seizures, mental retardation, microcephaly and other neurodevelopmental disorders), which aims to find disease causing mutations in a sample of 100 clients recruited from our Motor Speech Disorders Clinic at Department of Clinical Services, All India Institute of Speech and Hearing. This is a research project and hence ethical clearance was obtained, reference was mentioned in Methods. The client is a 15 year old female born out of a consanguineous union (parents were first cousins) diagnosed with microcephaly (occipitofrontal head circumference was 40 centimetres) and developmental delay. The mother had a history of one miscarriage at the third month of gestation and a still birth (female) at the eighth month (Figure 1A). An ultra sound scan of the foetus (client) at the eight month of pregnancy revealed delayed development. The client was born at term through normal delivery with no birth trauma. Her younger brother was clinically normal. The client had a squint at birth and had mild ID. At age 12 years, her developmental age was between 66 to 78 months as assessed by the Developmental Screening Test (DST)4. Her Receptive Language Age (RLA) and Expressive Language Age (ELA) was 18–20 months as revealed by Receptive Expressive Emergent Language Scale (REELS)5. She had a vocabulary of around 50 words and was able to comprehend commands, would recognize family members and common objects. She expressed herself through single word utterances, gestures and pointing.
Figure 1.
(A) The pedigree of the family; (B) workflow of the variant prioritization.
Sequencing methodology
The study was approved by the Institutional Ethical Body, All India Institute of Speech and Hearing [Ethical clearance reference number: SH/CDN/ARF-40/2016-17]. After obtaining written informed consent from the parents of the client, 5 ml of blood was collected from all family members (mother, father and brother) into EDTA coated vacutainers and DNA was isolated using Pure Link Genomic DNA Isolation Kit (Thermo Fisher Scientific), as per the manufacturer’s instructions. Approximately 100ng of genomic DNA was used to construct Exome libraries using Ion Ampliseq Exome RDY Panel (Thermo Fisher Scientific), as per the manufacturer’s protocol and these were quantified using High Sensitivity genomic DNA Assay on Qubit 3.0 (Thermo Fisher Scientific). Approximately 25 Pico moles of the library was used with the Ion Chef Instrument (Thermo Fisher Scientific) for template generation followed by, enrichment of templated ion sphere particles. Sequencing was performed using Hi-Q chemistry on Ion Proton system (Thermo Fisher Scientific) at our facility. Two samples were sequenced per chip and run generated 10.98 GB. The sample in question yielded 28,105,510 reads with 185 mean base pair length.
Results
For the client sample
QC filtered reads were aligned to reference genome GRCh37/hg19. Of the 28.1 million reads, 99.27% reads were on target with a mean genomic coverage of 91.83%. Mean depth was 81.32x with 89.87% mean uniformity. Variants were called from raw data using inbuilt variant caller plug-in present in Torrent suite (Version 5.2.2). Ion Reporter (version 5.4) annotated 38,085 variants from 13,013 genes to hg19 from the VCF file generated by variant caller plug-in. Variant prioritization was performed as shown in Figure 1B.
From the pedigree chart, we assumed autosomal recessive inheritance and filtered out heterozygous variants. This resulted in 75 homozygous variants from 67 genes. We found a novel frame shift insertion in exon 13 of the ASPM gene [(NM_018136.4), c.3384_3385 Insertion T], which induces a termination codon (p.Lys1129Ter) leading to non functional ASPM protein. Insertion was verified by Sanger sequencing using BDTv3 on 3500 Genetic Analyzer (Thermo Fisher Scientific). Homozygous insertion was confirmed in the client. Both parents and unaffected sibling were found to be heterozygous carriers (Figure 1A).
Discussion
ASPM protein determines cerebral cortical size. During initial stages of corticogenesis, the ASPM protein is essential in facilitating the proliferation of neural progenitors6. This process determines the cerebral cortical volume7 which has tripled over the last ~ 2 million years, leading to exceptionally big brain in humans compared to their primate counterparts8. This increase in the human brain size is believed to be one contributing factor for the emergence of higher cognitive function and language ability that are restricted to humans8. So far, 17 genes have been reported in which, mutations lead to the development of MCPH3,4,9–26. The phenotype(s) arising from pathogenic variants in these 17 genes are each named from MCPH 1 – MCPH 17 (there are 17 genes identified so far that cause autosomal recessive primary microcephaly, MCPH arising from these 17 genes are termed from MCPH 1 to MCPH 17) and the majority of the genetic load in MCPH is contributed by the ASPM gene, making MPCH5 the most prevalent of all the types of MCPH. Frame shift and protein truncating mutations in ASPM cause MCPH5 and these mutations are restricted to be seen in homozygous state only in the MCPH5 population27,28 (i.e., heterozygous mutations does not have any effect and only homozygous mutations will cause the MCPH).
Conclusion
The novel insertion mutation found in the ASPM gene in the present study segregated with the phenotype in the family, establishes the role of the novel frame shift mutation identifiedin the development of MCPH5 in the case studied. Candidate gene study by Sanger sequencing is time consuming and not economical when compared to WES. Given its higher diagnostic yield as evident by published studies on neurodevelopmental disorders2 and also from the present work, we support the findings reported by Rump et al. (2016) which states that WES in microcephaly population will end unnecessary further evaluations and aid in early appropriate interventions2.
Consent
Written informed consent to carry out the study and for the publication of the client’s and client’s sibling’s clinical details were obtained from the parents. Clinical details were obtained from the parents of the client.
Data availability
Sequence data for the insertion mutation (client sample) was deposited in Genbank under accession number MG063723.
Author contributions
SBD performed Exome Sequencing and Sanger sequencing, NS, NS and SK conceived the study, SV collected the data and its curation, SBD, SK performed data analysis and prepared manuscript, NS, NS supervised the work and corrected the draft, all the authors have read and approved the final manuscript.
Competing interests
No competing interests were disclosed.
Grant information
Grants to carry out the present project were provided by All India Institute of Speech and Hearing, an autonomous Institute under Ministry of Health and Family Welfare, Government of India, India.
Acknowledgements
The authors thank the participants for their cooperation. Our sincere thanks to Dr. S.R.Savithri, Director, All India Institute of Speech and Hearing, Mysore for permitting us to undertake the work.
Faculty Opinions recommended
References
1.
Rump P, Jazayeri O, van Dijk-Bos KK, et al.:
Whole-exome sequencing is a powerful approach for establishing the etiological diagnosis in patients with intellectual disability and microcephaly.
BMC Med Genomics.
2016; 9: 7. PubMed Abstract
| Publisher Full Text
| Free Full Text
2.
Abdel-Hamid MS, Ismail MF, Darwish HA, et al.:
Molecular and Phenotypic Spectrum of ASPM-Related Primary Microcephaly: Identification of Eight Novel Mutations.
Am J Med Genet Part A.
2016; 170(8): 2133–40. PubMed Abstract
| Publisher Full Text
3.
Hashmi JA, Al-Harbi KM, Ramzan K, et al.:
A novel splice-site mutation in the ASPM gene underlies autosomal recessive primary microcephaly.
Ann Saudi Med.
2016; 36(6): 391–396. PubMed Abstract
| Publisher Full Text
4.
Bharath Raj J:
DST Manual +Know your child’s intelligence and how to improve it. Sri Meera Printers: Mysore, 1983.
5.
Bzoch K, League R:
Receptive-Expressive Emergent Language (REEL) scale. Austin, TX:PRO-ED, 1971.
7.
Kouprina N, Pavlicek A, Mochida GH, et al.:
Accelerated Evolution of the ASPM Gene Controlling Brain Size Begins Prior to Human Brain Expansion.
PLoS Biol.
2004; 2(5): E126. PubMed Abstract
| Publisher Full Text
| Free Full Text
8.
Zhang J:
Evolution of the Human ASPM Gene, a Major Determinant of Brain Size.
Genetics.
2003; 165(4): 2063–2070. PubMed Abstract
| Free Full Text
9.
Jackson AP, Eastwood H, Bell SM, et al.:
Identification of microcephalin, a protein implicated in determining the size of the human brain.
Am J Hum Genet.
2002; 71(1): 136–142. PubMed Abstract
| Publisher Full Text
| Free Full Text
10.
Roberts E, Jackson AP, Carradice AC, et al.:
The second locus for autosomal recessive primary microcephaly (MCPH2) maps to chromosome 19q13.1-13.2.
Europ J Hum Genet.
1999; 7(7): 815–820. PubMed Abstract
| Publisher Full Text
11.
Moynihan L, Jackson AP, Roberts E, et al.:
A third novel locus for primary autosomal recessive microcephaly maps to chromosome 9q34.
Am J Hum Genet.
2000; 66(2): 724–727. PubMed Abstract
| Publisher Full Text
| Free Full Text
12.
Jamieson CR, Govaerts C, Abramowicz MJ:
Primary autosomal recessive microcephaly: homozygosity mapping of MCPH4 to chromosome 15.
Am J Hum Genet.
1999; 65(5): 1465–1469. PubMed Abstract
| Publisher Full Text
| Free Full Text
13.
Pattison L, Crow YJ, Deeble VJ, et al.:
A fifth locus for primary autosomal recessive microcephaly maps to chromosome 1q31.
Am J Hum Genet.
2000; 67(6): 1578–1580. PubMed Abstract
| Publisher Full Text
| Free Full Text
14.
Leal GF, Roberts E, Silva EO, et al.:
A novel locus for autosomal recessive primary microcephaly (MCPH6) maps to 13q12.2.
J Med Genet.
2003; 40(7): 540–542. PubMed Abstract
| Publisher Full Text
| Free Full Text
15.
Kumar A, Girimaji SC, Duvvari MR, et al.:
Mutations in STIL, encoding a pericentriolar and centrosomal protein, cause primary microcephaly.
Am J Hum Genet.
2009; 84(2): 286–290. PubMed Abstract
| Publisher Full Text
| Free Full Text
16.
Hussain MS, Baig SM, Neumann S, et al.:
A truncating mutation of CEP135 causes primary microcephaly and disturbed centrosomal function.
Am J Hum Genet.
2012; 90(5): 871–878. PubMed Abstract
| Publisher Full Text
| Free Full Text
17.
Jamieson CR, Govaerts C, Abramowicz MJ:
Primary autosomal recessive microcephaly: homozygosity mapping of MCPH4 to chromosome 15.
Am J Hum Genet.
1999; 65(5): 1465–1469. PubMed Abstract
| Publisher Full Text
| Free Full Text
18.
Yang YJ, Baltus AE, Mathew RS, et al.:
Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation.
Cell.
2012; 151(5): 1097–1112. PubMed Abstract
| Publisher Full Text
| Free Full Text
19.
Awad S, Al-Dosari MS, Al-Yacoub N, et al.:
Mutation in PHC1 implicates chromatin remodeling in primary microcephaly pathogenesis.
Hum Mol Genet.
2013; 22(11): 2200–2213. PubMed Abstract
| Publisher Full Text
20.
Hussain MS, Baig SM, Neumann S, et al.:
CDK6 associates with the centrosome during mitosis and is mutated in a large Pakistani family with primary microcephaly.
Hum Molec Genet.
2013; 22(25): 5199–5214. PubMed Abstract
| Publisher Full Text
21.
Mirzaa GM, Vitre B, Carpenter G, et al.:
Mutations in CENPE define a novel kinetochore-centromeric mechanism for microcephalic primordial dwarfism.
Hum Genet.
2014; 133(8): 1023–1039. PubMed Abstract
| Publisher Full Text
| Free Full Text
22.
Khan MA, Rupp VM, Orpinell M, et al.:
A missense mutation in the PISA domain of HsSAS-6 causes autosomal recessive primary microcephaly in a large consanguineous Pakistani family.
Hum Molec Genet.
2014; 23(22): 5940–5949. PubMed Abstract
| Publisher Full Text
23.
Alakbarzade V, Hameed A, Quek DQ, et al.:
A partially inactivating mutation in the sodium-dependent lysophosphatidylcholine transporter MFSD2A causes a non-lethal microcephaly syndrome.
Nat Genet.
2015; 47(7): 814–817. PubMed Abstract
| Publisher Full Text
24.
Guemez-Gamboa A, Nguyen LN, Yang H, et al.:
Inactivating mutations in MFSD2A, required for omega-3 fatty acid transport in brain, cause a lethal microcephaly syndrome.
Nat Genet.
2015; 47(7): 809–813. PubMed Abstract
| Publisher Full Text
| Free Full Text
25.
Yamamoto S, Jaiswal M, Charng WL, et al.:
A drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases.
Cell.
2014; 159(1): 200–214. PubMed Abstract
| Publisher Full Text
| Free Full Text
26.
Basit S, Al-Harbi KM, Alhijji SA, et al.:
CIT, a gene involved in neurogenic cytokinesis, is mutated in human primary microcephaly.
Hum Genet.
2016; 135(10): 1199–1207. PubMed Abstract
| Publisher Full Text
1
Unit for Human Genetics, All India Institute of Speech and Hearing, Manasagangothri, India 2
Department of Clinical Services, All India Institute of Speech and Hearing, Manasagangothri, India 3
Department of Speech Language Pathology, All India Institute of Speech and Hearing, Manasagangothri, India
Grants to carry out the present project were provided by All India Institute of Speech and Hearing, an autonomous Institute under Ministry of Health and Family Welfare, Government of India, India
Bhargav DS, Sreedevi N, Swapna N et al. Whole exome sequencing identifies a novel homozygous frameshift mutation in the ASPM gene, which causes microcephaly 5, primary, autosomal recessive [version 1; peer review: 2 approved]. F1000Research 2017, 6:2163 (https://doi.org/10.12688/f1000research.12102.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
The manuscript by Bhargav and colleagues describes a novel homozygous frameshift insertion mutation in a known microcephaly gene in a family segregating autosomal recessive primary microcephaly.
Whole exome sequencing was used to identify a mutation underlying MCPH phenotype.
... Continue reading
The manuscript by Bhargav and colleagues describes a novel homozygous frameshift insertion mutation in a known microcephaly gene in a family segregating autosomal recessive primary microcephaly.
Whole exome sequencing was used to identify a mutation underlying MCPH phenotype.
Overall, the manuscript is written well. Authors have presented the data in a professional way and I have
no reservation to approve this submission.
Is the work clearly and accurately presented and does it cite the current literature?
Yes
Is the study design appropriate and is the work technically sound?
Yes
Are sufficient details of methods and analysis provided to allow replication by others?
Yes
If applicable, is the statistical analysis and its interpretation appropriate?
Not applicable
Are all the source data underlying the results available to ensure full reproducibility?
Yes
Are the conclusions drawn adequately supported by the results?
Yes
References
1. Memon MM, Raza SI, Basit S, Kousar R, et al.: A novel WDR62 mutation causes primary microcephaly in a Pakistani family.Mol Biol Rep. 2013; 40 (1): 591-5 PubMed Abstract | Publisher Full Text 2. Kousar R, Hassan MJ, Khan B, Basit S, et al.: Mutations in WDR62 gene in Pakistani families with autosomal recessive primary microcephaly.BMC Neurol. 2011; 11: 119 PubMed Abstract | Publisher Full Text 3. Hashmi JA, Al-Harbi KM, Ramzan K, Albalawi AM, et al.: A novel splice-site mutation in the ASPM gene underlies autosomal recessive primary microcephaly.Ann Saudi Med. 36 (6): 391-396 PubMed Abstract | Publisher Full Text 4. Basit S, Al-Harbi KM, Alhijji SA, Albalawi AM, et al.: CIT, a gene involved in neurogenic cytokinesis, is mutated in human primary microcephaly.Hum Genet. 2016; 135 (10): 1199-207 PubMed Abstract | Publisher Full Text
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
NOTE: it is important to ensure the information in square brackets after the title is included in this citation.
Reviewer Report12 Jan 2018
Shahid Mahmood Baig,
National Institute for Biotechnology and Genetic Engineering, Pakistan Institute of Engineering & Applied Sciences, Faisalabad, Pakistan
In this report, a novel frameshift insertion in exon 13 of the ASPM gene in an Indian patient with autosomal recessive primary microcephaly has been reported.
This finding adds to the spectrum of mutations in ASPM gene using
... Continue reading
In this report, a novel frameshift insertion in exon 13 of the ASPM gene in an Indian patient with autosomal recessive primary microcephaly has been reported.
This finding adds to the spectrum of mutations in ASPM gene using whole exome sequencing (WES) for premarital screening and prenatal diagnosis to prevent affected births in at risk pregnancies. This report justifies the use of WES in delineating MCPH as compared to candidate gene approach. It is a descent short report, and hence recommended for indexing in its present form.
Is the work clearly and accurately presented and does it cite the current literature?
Yes
Is the study design appropriate and is the work technically sound?
Yes
Are sufficient details of methods and analysis provided to allow replication by others?
Yes
If applicable, is the statistical analysis and its interpretation appropriate?
Yes
Are all the source data underlying the results available to ensure full reproducibility?
Yes
Are the conclusions drawn adequately supported by the results?
Yes
Competing Interests: No competing interests were disclosed.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)