Keywords
Primary lymphedema, FOXC2, SNP, In silico, Bioinformatics, miRNA
Primary lymphedema, FOXC2, SNP, In silico, Bioinformatics, miRNA
We have followed the reviewers' notes, correcting the number of the total number of SNPs to 473, in addition to clarifying some points in the manuscript. As Dr Fengkai Zhang pointed out that the colours in the GeneMANIA network figure (Figure 3) are not clear, and because the colours cannot be changed on the online tool, we included the report provided by the online tool which outlines the type and strength of the interactions in addition to their related articles as Supplementary File 2. Also, a simplified text version of the network is available as Supplementary File 1. Supplementary Files 1 and 2 are unchanged from our version 1 paper.
To read any peer review reports and author responses for this article, follow the "read" links in the Open Peer Review table.
Lymphedema is an abnormal accumulation of interstitial fluid, due to inefficient uptake and reduced flow, leading to swelling and disability, which mostly affects the extremities. It can be divided into primary and secondary lymphedema according to the underlying cause. Primary or hereditary lymphedema results from genetic damage, while secondary or acquired lymphedema is caused by lymphatic system malfunction, resulting from trauma, including surgery, radiotherapy, tumors, or infections (for example, parasitic infections)1.
Hereditary lymphedema usually occurs as an autosomal dominant trait with allelic heterogeneity. The most common type of primary hereditary lymphedema, Milroy disease, can develop due to mutations in the vascular endothelial growth–factor receptor-3 gene (VEGFR-3; FLT4)2.
Forkhead box (Fox) proteins are a family of transcription factors (TFs) that play a key role in cell development, cell cycle regulation, and other important biological processes3. FOXC2, was first identified as a transcription factor (TF) that plays a key role in the morphogenesis of the cardiovascular system4. Further studies revealed that FOXC2 was involved in lymphatic vascular development. In both humans and mice, FOXC2 is expressed in large amounts in the developing lymphatic vessels and in adult lymphatic valves5. FOXC2-deficient mice were demonstrated to have abnormal lymphatic patterning and failure to form lymphatic valves, which reveals the critical role of FOXC2 in lymphatic vascular development6. Truncating and missense mutations of FOXC2 have been discovered in patients with late-onset lymphedema (hereditary lymphedema II; OMIM 153200), often associated with distichiasis (double row of eyelashes), and sometimes ptosis (Lymphedema Distichiasis Syndrome [LDS]; OMIM 153400), and/or yellow nails (OMIM 153300)7–9. LDS patients develop defects characterized by lymphatic and venous blood reflux, which means failure and/or absence of lymphatic and venous valves10,11. On a molecular level, FOXC2 DNA binding sites are enriched in nuclear factor of activated T-cells 1 (NFATC1) consensus sequences, and the two TFs work in tandem during lymphatic vascular morphogenesis12. FOXC2 controls the expression of proteins that are essential for lymphatic valve development, such as connexins. Thus, adequate control of their activity is extremely important for proper lymphatic vessel development and function13.
The aetiology governing the phenotypic variability remains unclear14. Lymphatic malformations could also result from slightly mutated germline alleles, which are difficult to access (and so to identify), mutations in regulatory regions of the DNA, epigenetic changes, or a combination of the three. Epigenome sequencing and whole exome sequencing may be needed for in-depth study of these mutations. The part genetics play in the development of lymphatic anomalies is highly complex, shown by the discovery of 23 mutated human genes13.
Few studies addressing FOXC2 mutations from a bioinformatics point of view have been published. Out of those, none have specifically focused on single nucleotide polymorphisms (SNPs). SNPs may affect codons of amino acids located at the forkhead active domain of the protein or other sites and may severely affect the function of the TFs. Furthermore, SNPs are feasible and cost effective to study using in silico analysis via available bioinformatics tools.
This study aimed to analyze all SNPs in the human FOXC2 gene and predict their effect on the structure, function, stability and regulation of its respective protein. The results of our study can be used in population studies to screen patients with hereditary lymphedema, and more importantly in phenotypic variations in lymphatic malformations of affected individuals.
We selected the National Center for Biotechnology Information (NCBI) database, dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP) for the retrieval of SNPs and their related protein sequence of FOXC2 gene. We used “FOXC2” as our search term and used filters to narrow down our search results into two categories: 3’ + 5’ UTR SNPs only and all SNPs in FOXC2, except for those in the 3’ + 5’ UTR. This gene was chosen because it’s the one that is known to be associated with LDS, used for our computational analysis. It should be noted that dbSNP has its caveats, like high false positive rates, but this was partially overcome by using several SNP effect prediction tools (as outlined below).
We chose three complementary algorithms for functional impact prediction of nsSNPs: Sorting Intolerant From Tolerant (SIFT; http://sift.bii.a-star.edu.sg/), Polymorphism Phenotyping (PolyPhen; http://genetics.bwh.harvard.edu/pph/) and CONsensus DELeteriousness (Condel; http://bg.upf.edu/fannsdb/)15–17.
SIFT version 2.0 was used to distinguish between tolerant and intolerant coding mutations, and is used to predict whether an amino acid substitution in a protein will have a phenotypic effect. SIFT is based on the premise that protein evolution is correlated with protein function. Variants that occur at conserved alignment positions are expected to be tolerated less than those that occur at diverse positions. PolyPhen is a computational tool for identification of potentially functional nsSNPs. Predictions are based on a combination of phylogenetic, structural and sequence annotation information characterizing a substitution and its position in the protein. These algorithms can possibly identify pathogenic SNPs, and using three algorithms increases the robustness of the results by avoiding false negatives and positives.
We uploaded the SNP accession numbers of all the SNPs into VEP (http://www.ensembl.org/Tools/VEP)18 and enabled “SIFT”, “PolyPhen” and “Condel” to retrieve the corresponding predictions of the functional significance of each SNP from all of the three algorithms. We repeated this step using Biomart (http://www.ensembl.org/biomart/martview/)19, which is similar to VEP and part of Ensembl. It provides a SIFT and PolyPhen prediction and score, like VEP, but it does not provide a Condel prediction. We took the SNPs predicted by both databases to be deleterious and pathogenic (2 SNPs; rs121909106 and rs121909107) to perform the next steps in the analysis.
Project HOPE (http://www.cmbi.ru.nl/hope/) is an online web-server used to search protein 3D structures by collecting structural information from a series of sources. We entered the two SNPs (rs121909106 and rs121909107) that we obtained from the last step together with the primary structure of the FOXC2 protein (obtained from the Protein Data Bank http://www.rcsb.org/pdb/explore/explore.do?structureId=1D5V) into HOPE20. HOPE lets you choose the amino acid that was affected by the mutation and allows you to change that specific amino acid and then analyse the resulting change in the 3D (tertiary/quaternary) structure and outputs the change predicted, plus the explanation of such a change (in both structure and function of the protein).
Gene-gene interactions were studied to highlight candidate genes that could possibly be associated with LDS, especially if haplotypes were to be studied in the future. GeneMANIA ver. 3.1.2.8 (http://genemania.org/) finds other genes that are related to a set of input genes, using a very large set of functional association data21. It was chosen for its speed and accuracy in prediction of gene-gene interactions, in addition to its relatively frequent version updates. Association data include protein and genetic interactions, pathways, co-expression, co-localization and protein domain similarity. We entered FOXC2 as our query gene and the website generated a network of genes along with their gene-gene interactions, according to gene ontology terms.
PolymiRTS v3.0 (http://compbio.uthsc.edu/miRSNP/) is an integrated platform for analyzing the functional impact of genetic polymorphisms in miRNA seed regions and miRNA target sites22. We entered a second set of SNPs (containing only 5’ and 3’ SNPs) and acquired a list of the miRNAs affected by these mutations. The affected miRNAs might lead to a decrease/increase of the expression of FOXC2.
The analysis steps performed are summarized in Figure 1.
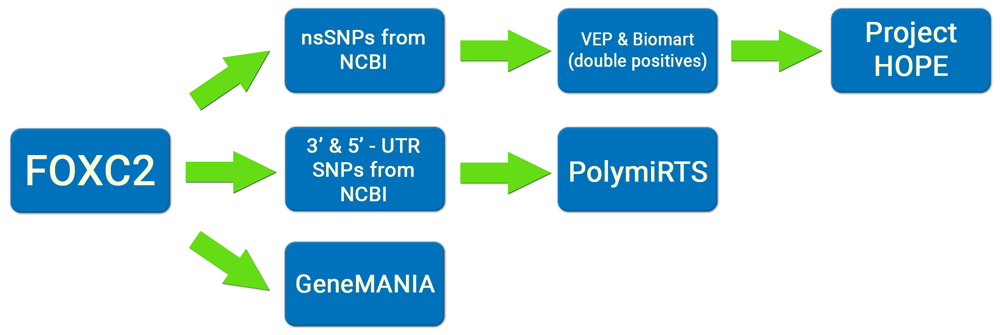
SNP information for FOXC2 was retrieved from dbSNP. For our investigations, we selected SNPs in coding and UTR (5’and 3’) regions. Among the 473 SNPs, 429 were nsSNPs and 44 SNPs were in the 5’ and 3’ UTRs of FOXC2.
We found two SNPs, rs121909106 and rs121909107, which correspond to S125L and R121H, to have significant deleterious effect on the structure and function of FOXC2 protein. We summarized the information of the two SNPs predicted by the two databases in Table 1.
Figure 2 shows a 3D model of rs121909107 and rs121909106 and the spatial effects of each on the respective domains they are a part of, in addition to their effects on neighbouring domains.
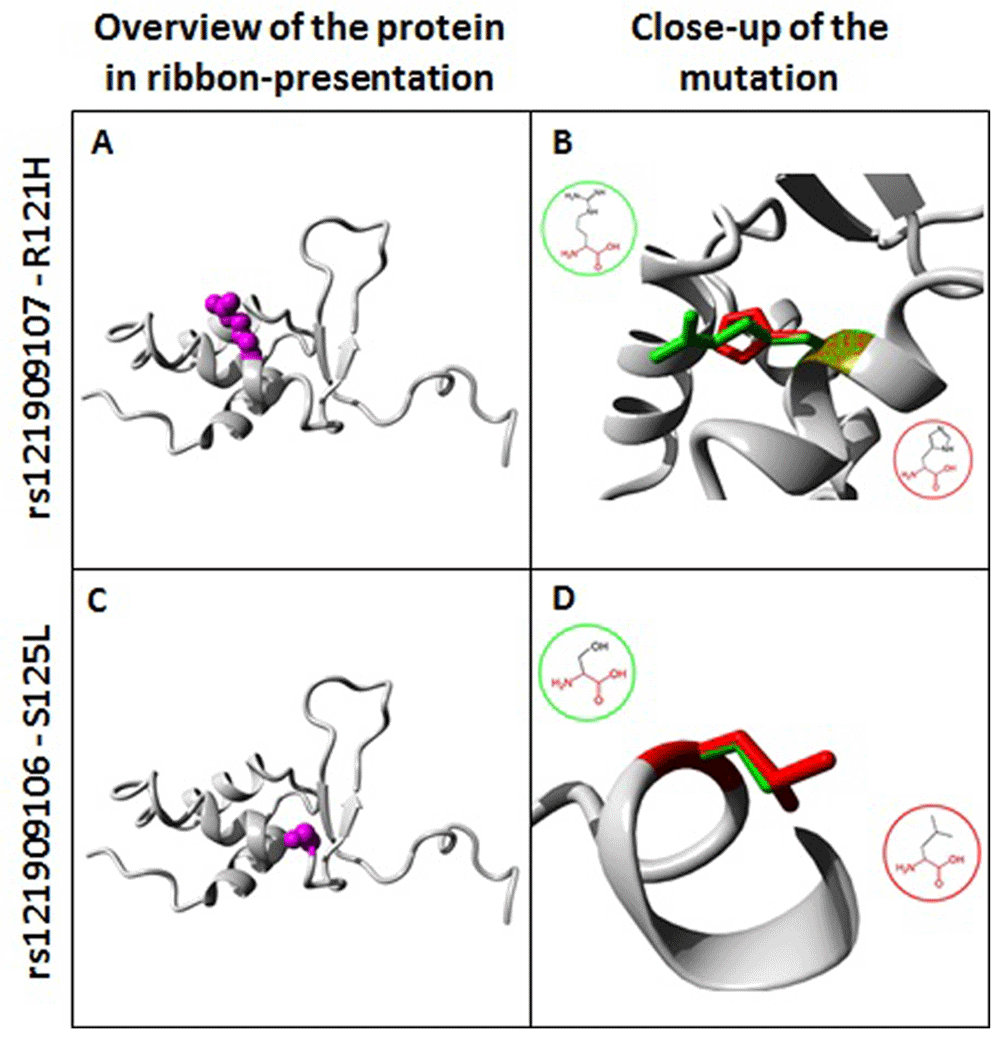
Overview shows the protein in grey and the affected amino acid in purple, the close-up shows the side chains of both the wild-type (Arginine) and the mutant residue (Histidine) at position 121 and colored green and red respectively. For rs121909106; overview shows the protein in grey and the affected amino acid in purple, the close-up shows the side chains of both the wild-type (Serine) and the mutant residue (Leucine) at position 125 are shown and colored green and red, respectively.
Figure 3 shows the gene-gene interactions of FOXC2. The most important interactions are with FOXB1, FOXD1, FOXD2 and FOXD3 (Figure 3) (additional data in Supplementary material).
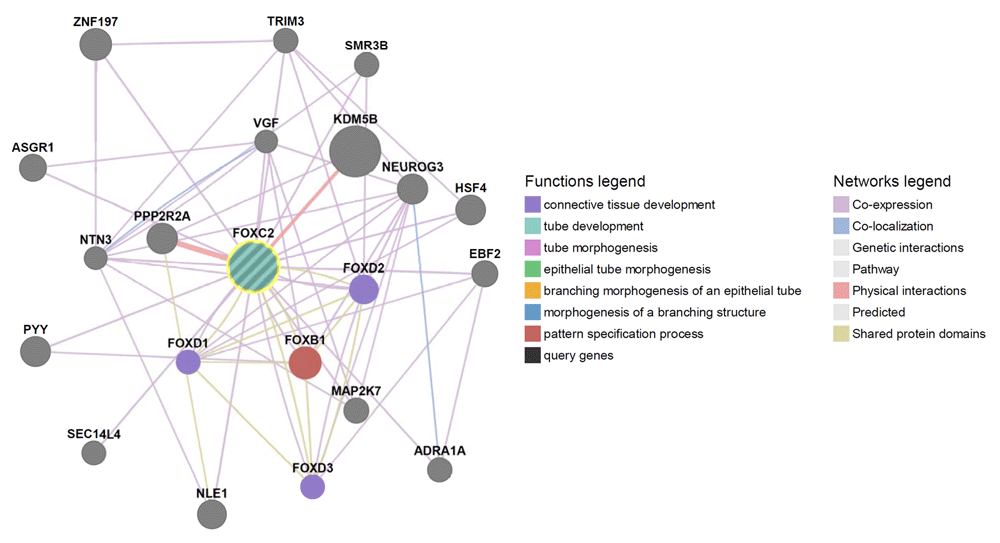
Table 2–Table 4 show the effect that SNPs in the 3’ and 5’ region of FOXC2 might have, along with the conservation score (CS) and context+ score of the site in question. The higher the CS, the more profound the effect of the SNP is predicted to be. In addition, the higher the context+ score, the higher the likelihood of a change (disruption or creation) occurring in the miRNA target site.
| Location | dbSNP ID | Wobble base pair | Ancestral allele | Allele | miR ID | Conservation | miRSite | Function Class | Exp Support | context+ score change |
|---|---|---|---|---|---|---|---|---|---|---|
| 86602454 | rs200353178 | N | C | C | hsa-miR-1247-3p hsa-miR-4449 hsa-miR-4532 | 2 3 3 | cgtgTCCCGGGAc cgtgtcCCGGGAC cgtgtCCCGGGAc | D D D | N N N | No Change No Change No Change |
| T | hsa-miR-4740-3p | 2 | cgtgTCTCGGGAc | C | N | No Change | ||||
| 86602489 | rs3751795 | N | C | C | hsa-miR-4747-5p hsa-miR-5196-5p | 2 2 | gcttcgCTTCCCA gcttcgCTTCCCA | D D | N N | -0.054 -0.057 |
| T | hsa-miR-3153 hsa-miR-4668-5p hsa-miR-6733-5p hsa-miR-6739-5p | 2 2 2 2 | gcttcgTTTCCCA gcttcgTTTCCCA gcttcgTTTCCCA gcttcgTTTCCCA | C C C C | N N N N | -0.024 -0.022 -0.021 -0.027 | ||||
| 86602513 | rs142773766 | N | C | C | hsa-miR-4288 hsa-miR-632 | 2 2 | acCAGACAAttaa acCAGACAAttaa | D D | N N | -0.117 -0.117 |
| T | hsa-miR-1298-3p hsa-miR-3126-3p | 3 3 | aCCAGATAattaa aCCAGATAattaa | C C | N N | -0.114 -0.132 | ||||
| Location | SNP location in the mRNA transcript. It is a zero-based number. | |||||||||
| SNPID | Link to dbSNP. | |||||||||
| Wobble Pair | Whether the SNP can form a G:U wobble basepair with the miRNA. Y: Yes; N: No. | |||||||||
| Ancestral Allele | If applicable, the ancestral allele is denoted. | |||||||||
| Allele | Two alleles of the SNP in the mRNA transcript. | |||||||||
| miRID | Link to miRBase. | |||||||||
| Conservation | Occurrence of the miRNA site in other vertebrate genomes in addition to the query genome. By clicking the hyperlink, the users can examine the genomes in which this miRNA target site occurs. | |||||||||
| FuncClass | D: The derived allele disrupts a conserved miRNA site (ancestral allele with support >= 2). N: The derived allele disrupts a nonconserved miRNA site (ancestral allele with support < 2). C: The derived allele creates a new miRNA site. O: The ancestral allele can not be determined. | |||||||||
| miRSite | Sequence context of the miRNA site. Bases complementary to the seed region are in capital letters and SNPs are highlighted in red. | |||||||||
| ExpSupport | LT: The miRNA-mRNA interaction is supported by a low-throughput experiment (e.g., luciferase reporter assay or Western blot). HT: The miRNA-mRNA interaction is supported by a high-throughput experiment (e.g., microarray or pSILAC). LTL: The miRNA targeting the specific location is supported by a low-throughput experiment (e.g., allelic imbalance sequencing). HTL: The miRNA targeting the specific location is supported by a high-throughput experiment (e.g., HITS-CLIP). | |||||||||
| Context+ score change | Context+ scores predict the binding of a miRNA to the entire 3'-UTR by summing over contributions made by individual sites within the 3'-UTR that have perfect sequence complementarity to the miRNA seed region. The change the differences in context+ scores between the reference and derived alleles for each SNP or INDEL in putative miRNA target sites. A more negative value of the context+ score difference indicates an increased likelihood that the miRNA targeting is disrupted or newly created by the mutation in the target sites | |||||||||
| Location | miR ID | dbSNP ID | miR seed | Allele | Wobble base pair | miRSite | Conservation | context+score change |
|---|---|---|---|---|---|---|---|---|
| 86602466 | hsa-miR-6889-5p | rs146254801 | C[G/A]GGGAG | G/A | 1 | CUCCCCG | 2 | -0.182 |
| 86602496 | hsa-miR-5090 | rs3823658 | C[G/A]GGGCA | G/A | 1 | GCCCCGA | 2 | -0.227 |
| 86602496 | hsa-miR-6727-5p | rs202175375 | U[C/T]GGGGC | C/T | 0 | GCCCCGA | 2 | -0.291 |
| 86602466 | hsa-miR-6777-5p | rs56155608 | [C/T]GGGGAG | C/T | 0 | CUCCCCG | 2 | -0.144 |
| Location | miR ID | dbSNP ID | miR Seed | Allele | Wobble base pair | miRSite | Conservation | context+score change |
|---|---|---|---|---|---|---|---|---|
| 86602524 | hsa-miR-6886-5p | rs201118690 | [C/T]CGCAGG | C/T | 0 | CUGCAGA | 7 | -0.074 |
| 86602524 | hsa-miR-6886-5p | rs6413505 | C[C/T]GCAGG | C/T | 0 | CUGCAGA | 7 | -0.074 |
| 86602530 | hsa-miR-6720-3p | rs201914560 | GCG[C/T]CUG | C/T | 0 | AGACGCA | 6 | -0.108 |
| 86602497 | hsa-miR-4472 | rs28655823 | GU[G/C]GGGG | G/C | 0 | CCCCGAC | 2 | -0.244 |
| 86602497 | hsa-miR-4472 | rs202127912 | GU[TG/-]GGGG | TG/- | 0 | CCCCGAC | 2 | -0.244 |
This study aimed to analyze the SNPs identified in the FOXC2 gene. We found a total of 473 SNPs, 2 of which were predicted to adversely affect the function of the resulting protein. One mutation leads to translation of a histidine instead of an arginine at position 121, and the mutant residue is located near a highly conserved region20. The mutation was reported previously in a patient suffering from lymphedema-distichiasis syndrome by Berry et al. (2005), and the effects that the R121H mutation would have on the DNA-binding part of the forkhead domain (FHD) of FOXC2 was predicted. It was predicted that R121H would impair FOXC2 protein’s ability to bind DNA and act as a transcription activator, and this was confirmed by biochemical studies. Also, the mutation resulted in the mislocalization of FOXC2. Berry et al. (2005) determined that the mutation results in a non-functional protein and that this leads to hereditary LDS23. The residue affected by rs121909107 is part of an inter pro-domain named Fork Head Domain Conserved Site 2 (Interpro, IPR030456), and it is annotated with the following Gene-Ontology (GO) terms to indicate its function: sequence-specific DNA binding (GO:0043565) and sequence-specific DNA binding transcription factor activity (GO:0003700).
The change of a serine into a leucine at position 125 means that a nonpolar amino acid will be replaced with a polar one. The original wild-type residue and newly introduced mutant residue differ in their electrochemical properties. The mutation results in incorporating an amino acid with a different level of hydrophobicity, this will affect hydrogen bond formation, and the mutant residue is located near a highly-conserved region20. This mutation matches a previously described variant in affected members of families with lymphedema-distichiasis syndrome, previously reported by Mangion et al.24 and Bell et al.7. The mutation was not identified in 100 normal chromosomes. This mutation results in a wide range of phenotypes from minimal distichiasis to severe lymphoedema and congenital heart disease7. The residue affected by rs121909106 is part of an inter prodmain named Winged Helix-Turn-Helix DNA-Binding Domain (IPR011991) and it is annotated with the following GO terms to indicate its function: nucleic acid binding (GO:0003676) and nucleic acid binding transcription factor activity (GO:0001071). No coordination was found between the two SNPs, and they are not known to belong to any haplotype.
Our results from GeneMANIA showed that FOXC2 interacts with a lot of genes (Figure 3), mainly functioning to control connective tissue, tube and epithelial tissue development. These results were proved important by Mangion et al.24 and Bell et al.7, which showed that mutations in FOXC2 lead to developing LDS.
PolymiRTS results showed SNPs and INDELs in miRNA target sites: target sites disrupted by SNPs and INDELs in miRNA seeds and target sites created by SNPs and INDELs in miRNA seeds (Table 2–Table 4). Three miRNAs that are worth noting are hsa-miR-6886-5p (with a CS of 7), hsa-miR-6886-5p (with a CS of 7) and hsa-miR-6720-3p (with a CS of 6), which are affected by the SNPs rs201118690, rs6413505, rs201914560, respectively. This points to the possibility that the areas affected by those SNPs have an evolutionary important function.
In conclusion, there are many SNPs that affect FOXC2 gene; some are predicted to be harmful, such as rs121909106 and rs121909107, but most are not. miRNAs were affected by SNPs in the 3’ and 5’ untranslated regions of FOXC2 gene and three are noteworthy, hsa-miR-6886-5p, hsa-miR-6886-5p and hsa-miR-6720-3p, due their high conservation score.
Computational biology tools are very powerful, especially when provided with good data and used by experts. However, bioinformatics tools have their limitations; most importantly the fact that their results are but predictions, meaning that the information they provide us with requires confirmation using various methods such as functional studies.
Dataset 1: SNPs used for PolymiRTS analysis. doi, 10.5256/f1000research.10937.d15306425
Dataset 2: SNPs used for other analyses. doi, 10.5256/f1000research.10937.d15306526
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)