Introduction
α-synuclein is a small, highly abundant, and highly conserved presynaptic protein with intimate links to many neurodegenerative diseases and is one of the more intensively studied human proteins. In fact, since its discovery in 19911, followed by the demonstration of its natively unfolded nature2, and especially after finding in 1997 a potential relationship between α-synuclein aggregation and the pathology of Parkinson’s disease (PD)3,4, this protein has attracted the close attention of many researchers specializing in various scientific areas. According to the Web of Science database, as of 17 February 2017, there were 15,367 publications about this protein, reflecting collective efforts of 37,066 researchers affiliated with 6,326 organizations from 93 countries/territories, and more than 3,140 papers were published on α-synuclein during the past 2 years alone. Figure 1 represents a more detailed view of these Web of Science data by showing the cumulative number of publications for the past 26 years and the annual number of publications dedicated to this protein.
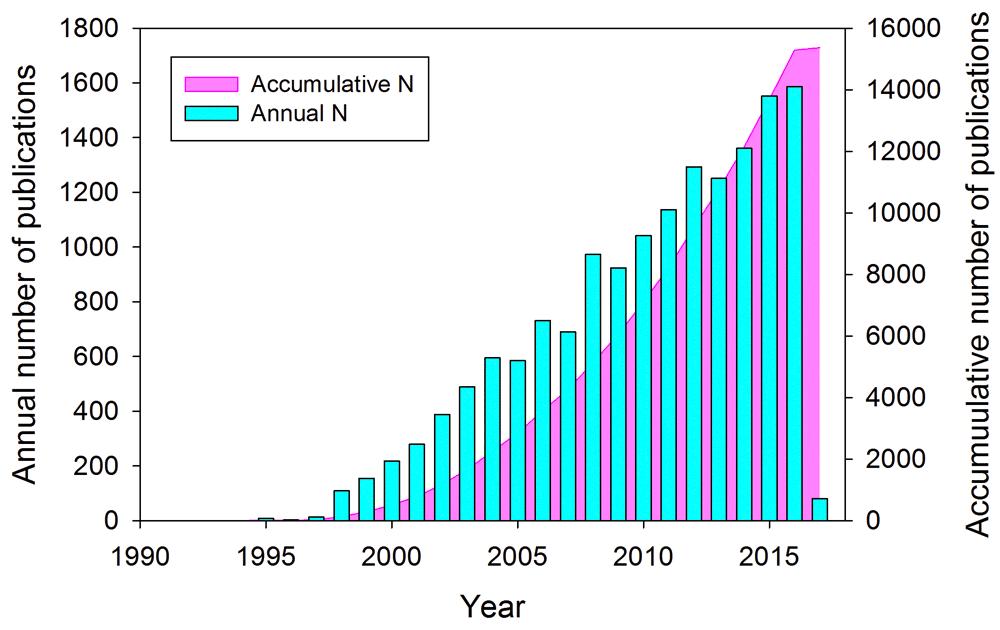
Figure 1. A time course of the development of interest in α-synuclein-related research.
Web of Science data related to the publications dedicated to α-synuclein: the cumulative number of publications for the past 26 years (pink area plot) and the annual number of publications dedicated to this protein (cyan bars).
Despite immense efforts, this 25-year-old enigmatic protein continues to keep multiple secrets related to its structure, function, dysfunction, multipathogenicity, and pathology transmission. The aforementioned considerations constitute a perfect stage for an important question related to α-synuclein: what is so special about this protein that makes it a multipathogenicity carrier? The goal of this article is to shed some light on α-synuclein-related mysteries by presenting the most recent advances in our understanding of this protein and its aggregation.
Multipathogenicity angle
To better understand why researchers continue to study this protein, let’s consider what multipathogenicity means for α-synuclein. Although originally considered a potential cause of PD (where aggregated α-synuclein is present in the form of intracellular inclusions, Lewy bodies [LBs] and Lewy neurites [LNs]5,6), this protein is known to be involved in the pathogenesis of a diverse group of neurodegenerative diseases collectively known as synucleinopathies. Some of these maladies include Alzheimer’s disease (AD), neurodegeneration with brain iron accumulation type 1, pure autonomic failure, Down’s syndrome, amyotrophic lateral sclerosis-parkinsonism-dementia complex of Guam, multiple system atrophy (MSA), and several LB disorders (that, in fact, might represent a clinical continuum7), such as sporadic and familial PD, dementia with LBs (DLB), diffuse LB disease, the LB variant of AD, and PD dementia4,8–15. These (and potentially many other) neurodegenerative diseases can be considered α-synuclein-related brain amyloidoses, since all of them are characterized by the presence of common pathological intracellular inclusions containing α-synuclein in selectively vulnerable neurons and glia and since the onset and progression of their clinical symptoms, as well as the degeneration of affected brain regions, are linked to the formation of abnormal filamentous aggregates containing α-synuclein9,15–21. Importantly, accumulated evidence indicates that α-synuclein-based aggregation and deposition of LBs can affect multiple areas of the peripheral and central nervous systems, such as the dorsal raphe nucleus, dorsal nucleus of the vagus nerve, hypothalamic nuclei, intermediolateral nucleus, locus coeruleus, nucleus basalis of Meynert, and substantia nigra22, whereas LNs can be found in the basal ganglia, cerebral cortex, dorsal nucleus of the vagus nerve, and sympathetic ganglia as well as in the intramural autonomic ganglia of the gastrointestinal tract23,24. In line with these earlier observations, a recent study of the inclusion pathologies in PD and DLB confirmed the abundant presence of LBs and LNs in the dorsal motor vagal and solitary nuclei, locus coeruleus, parabrachial nuclei, pedunculopontine and raphe nuclei, periaqueductal gray, prepositus hypoglossal, substantia nigra, reticular formation, and ventral tegmental area and demonstrated the presence of LN in brainstem fiber tracts and the existence of LBs and LNs in cranial nerve, premotor oculomotor, precerebellar, and vestibular brainstem nuclei25. This study also supported an important notion that α-synuclein deposition-related pathological processes can spread and do so transneuronally along anatomical pathways25. Recently, application of the α-synuclein proximity ligation assay revealed the presence in the post-mortem brain tissue from PD patients of previously unrecognized pathology in the form of extensive diffuse deposition of α-synuclein oligomers that were often localized, in the absence of LBs, to neuroanatomical regions mildly affected in PD26.
Obviously, although the diversity of synucleinopathies and related symptoms potentially can be attributed to the complexity of the organization of brain and nervous system (different motor and non-motor symptoms are the manifestation of the malfunction of different brain and nervous system regions), this complexity, in general, cannot be used to explain the cause of the multifaceted, α-synuclein-related pathology at the molecular level. In fact, since α-synuclein is highly expressed throughout the brain, accounting for as much as 1% of the total protein in soluble cytosolic brain fractions27, and since any given synucleinopathy possesses unique spatiotemporal characteristics (happens at a specific time due to the malfunction of a specific brain region), some other factors, likely related to α-synuclein and its interaction with the environment (such as mutations in the SYNC gene, presence of alternatively spliced isoforms, post-translational modifications (PTMs), toxic insult, oxidative stress, presence of dopamine, metal ions, specific binding partners, etc.), should be taken into account.
Connection of synucleinopathies to other diseases angle
Recent studies provided further support for an interesting molecular link between the synucleinopathies and other neurodegenerative diseases by demonstrating the ability of α-synuclein to interact with and regulate proteins specific to several degenerative maladies. For example, it was shown that α-synuclein can interact and co-aggregate with mutant huntingtin, a protein related to Huntington's disease28, and with tau protein, the aggregation of which is associated with various tauopathies, including AD29. A cooperation of α-synuclein with β-amyloid (Aβ) was shown to block SNARE-dependent vesicle fusion30, whereas interaction with Aβ led to the inhibition of Aβ deposition and reduced plaque formation31. Finally, α-synuclein was also shown to engage in interaction with autophagy/beclin1 regulator 1, a protein related to MSA pathogenesis, and this binding was dramatically enhanced by the α-synuclein phosphorylation at serine 12932. Note that the aforementioned interactions of α-synuclein with the proteins associated with other neurodegenerative diseases can be grouped into two classes – interactions leading to the co-aggregation of α-synuclein with said proteins and interactions leading to the modulation of functionality of proteins targeted by α-synuclein.
Multifunctionality angle
Many papers in the field start with an introductory sentence stating that α-synuclein is a small, highly conserved presynaptic protein with unknown function. This statement is a bit odd, taking into account all the efforts of numerous researchers working on α-synuclein. In fact, according to the PubMed database (as of 16 December 2016), there were more than 7,150 papers mentioning synuclein function, many of those papers were dedicated to the detailed investigation of what this protein can do, and, as a result, many potential functions were ascribed to α-synuclein. The explanation of this contradiction (many functions are described, but function is unknown) is in the logistics of the classical structural biology relying on the influential “one gene – one enzyme – one function” hypothesis, according to which each gene encodes a single protein that has a unique biological function and is responsible for a single step in a metabolic pathway33. In line with this hypothesis, distortion of normal protein function (in the form of loss of a normal function or gain of a pathological function) might represent the molecular basis of a proteinopathy.
However, since α-synuclein was shown to have not one but many functions (see below), this immediately brought significant uneasiness to functional data interpretation, leading to the logical conclusion that the observed multifunctionality indicates the lack of a unique function and, therefore, could be unreal. Although, from the viewpoint of the “one protein – one function” model, these observations provide grounds for the “protein with unknown function” epithet, an alternative (and much more intriguing and lucrative) hypothesis stating that α-synuclein is indeed a multifunctional protein, which can, in fact, do everything ascribed to it (and, probably, much more than that). In this case, problems with different α-synuclein-based functions might be directly or indirectly related to the pathogenesis of different synucleinopathies. In other words, a spectrum of functions can cause a spectrum of dysfunctions that might lead to a spectrum of diseases.
Early work in this direction showed that functions of α-synuclein can range from fatty acid binding34 to interaction with plasma membranes and formation of membrane channels or modification of their activity35, association with mitochondria causing mitochondrial dysfunction35, metal binding36–39, interaction with pesticides and herbicides40–42, synaptic vesicle release and trafficking34, positive and negative regulation of neurotransmitter release43, regulation of certain enzymes and transporters34, control of neuronal survival34, regulation of the neuronal apoptotic response44 and protection of neurons from various apoptotic stimuli44, and promiscuous interaction with hundreds of unrelated proteins and other binding partners34,45–47. In the past 2 years, the phenomenon of α-synuclein multifunctionality was further confirmed and elaborated by adding a long list of new functions. Note that the inventory below is far from being exhaustive, since over 1,100 articles dedicated to synuclein function were published during the last 2 years, and, therefore, it is physically impossible to cover all new developments in this commentary owing to the space restrictions.
Knockout studies of the synuclein functionality
Knockout experiments, where the target gene is made inoperative, represent a useful approach for the evaluation of the global biological role of a query protein. Earlier studies showed that although α-synuclein-null mice were viable, fertile, and characterized by intact brain architecture, they possessed altered dopamine release and displayed a reduction in striatal dopamine and attenuated dopamine-dependent locomotor response48. Furthermore, α-synuclein-null mice were characterized by a selective deficiency of undocked vesicles without affecting docked vesicles in hippocampal synapses49, showed abnormal compartmentalization of norepinephrine in dentate gyrus50, possessed greatly increased rates of operant behavior during intracranial self-stimulation51, and showed an earlier onset of symptoms of experimental autoimmune encephalomyelitis52 but were resistant to the Parkinsonian neurotoxin MPTP that inhibits mitochondrial complex I53. This MPTP resistance was also present in the γ-synuclein and double α-synuclein/γ-synuclein-null mutant animals54, and α-synuclein-null mice were shown to have an attenuated loss of striatal dopamine caused by prolonged chronic MPTP administration55.
Multiple studies of the α-synuclein-knockout models suggested that this protein plays a role in lipid metabolism. In fact, α-synuclein-null animals were characterized by decreased brain palmitate uptake and altered palmitate metabolism56, showed increased incorporation and turnover of the docosahexaenoic acid in brain phospholipids57, possessed reduced arachidonate turnover in brain phospholipids58, and showed increased mass of brain neutral lipids59.
Binding promiscuity
Among biological activities recently ascribed to α-synuclein is a numerous set of examples providing solid support for its reputation as a promiscuous binder. In fact, multiple new examples were reported showing that this protein is engaged in interaction with the multifunctional co-chaperone Bcl-2-associated athanogene-160, prolyl oligopeptidase leading to enhanced α-synuclein dimerization61, various synaptosomal proteins62, mitochondria-associated membranes63, molecular chaperone Munc18-1, which is a key component of the exocytic machinery that controls the release of neurotransmitters64, the neuronal phosphoprotein synapsin III65, voltage-dependent anion channel66, a suicide inhibitor of monoamine oxidases, rasagiline67, and the aldehyde of serotonin, 5-hydroxyindoleacetaldehyde68.
Control of cellular processes
Furthermore, α-synuclein has been linked to several cellular processes, such as activation of microglia69, involvement in the regulation of autophagy70,71, initiation of innate and adaptive immune responses72, membrane remodeling73,74, regulation of synaptic vesicle size75, contribution to axonal transport impairment76, stress-induced mitochondrial morphological remodeling (in cooperation with parkin and PINK1)77, alteration of endoplasmic reticulum–mitochondrial communication78, promotion of microtubule nucleation and enhancement of microtubule growth rate via interaction with microtubules and tubulin α2β2 tetramer79, and inhibition of signaling related to the ATF6 pathway, which is a protective branch of the unfolded protein response80, and even serving as a brain antimicrobial peptide that exhibits noticeable antibacterial activity against Escherichia coli and Staphylococcus aureus81.
α-synuclein radicals
Recently, Kumar et al. exploited an intriguing possibility that the formation of proteinaceous radicals might contribute to PD pathogenesis82. Using the highly sensitive immuno-spin trapping technique, the authors showed that in the midbrains of maneb- and paraquat-co-exposed mice, the activation of NADPH oxidase and inducible nitric oxide synthase took place, eventually leading to the peroxynitrite-mediated formation of α-synuclein radicals in the dopaminergic neurons of exposed mice82. Furthermore, the process of α-synuclein radical formation paralleled death dopaminergic neurons in the midbrains of maneb- and paraquat-co-exposed mice, indicating that there is an intricate link between protein radicals and disease progression82.
Regulation of various proteins
α-synuclein was also shown to regulate complexin-1 and midbrain-specific factor forkhead box P1 expression83, enhance histone H3 lysine-9 (H3K9) dimethylation and promote H3K9me2-dependent transcriptional responses84, protect the functions of Hsp90 clients, such as Akt and mTOR, when the activity of Hsp90 is blocked85, stimulate protein phosphatase-2A (PP2A) activity and regulate tyrosine hydroxylase phosphorylation via the control of PP2A methylation86, modulate the trafficking and function of glutamate N-methyl-d-aspartate receptor87, and promote Notch1 intracellular domain degradation via interaction with the ubiquitin E3 ligase Fbw788.
α-synuclein in the family circle
Synucleins constitute a family of closely related presynaptic proteins encoded by three distinct genes and are found only in vertebrates89. In addition to α-synuclein, which is also known as the non-amyloid-β component (NAC) precursor protein or synelfin90–92, this family includes β-synuclein (phosphoneuroprotein 14 or PNP14)92–94, and γ-synuclein (breast cancer-specific gene 1 [BCSG1] or persyn)95–98. There is 78% identity between human α- and β-synucleins, with the major difference between the two being the lack of the significant part of the NAC region13,99. Human γ-synuclein shares 60% sequence similarity with α-synuclein and lacks the tyrosine-rich C-terminal tail that represents a signature of α- and β-synucleins13,99. Importantly, in addition to the traditional α-synuclein-containing pathological inclusions, the development of several synucleinopathies is accompanied by the appearance of α-, β-, and γ-synuclein-positive vesicular-like lesions at the presynaptic axon terminals in the hippocampal dentate, hilar, and CA2/3 regions100.
Structurally, all members of the synuclein family are intrinsically disordered at physiological conditions but adopt comparable partially folded conformations at acidic pH or at high temperature101. Both α- and γ-synucleins can easily form fibrils under a variety of identical conditions101, whereas β-synuclein fibrillation requires the presence of some metal ions (Zn2+, Pb2+, and Cu2+)102.
Fibrillation of human α-synuclein can be inhibited by the addition of either β- or γ-synuclein in vitro101, with β-synuclein being shown to inhibit α-synuclein aggregation in an animal model103. NMR paramagnetic relaxation enhancement experiments revealed that β-synuclein forms transient heterodimers with α-synuclein that are characterized by high specificity and weak affinity104. Recently, the competition of β-synuclein for binding sites at the surfaces of lipid vesicles and at the surfaces of α-synuclein fibrils was proposed as a molecular mechanism of the β-synuclein-driven inhibition of lipid-induced α-synuclein aggregation and secondary nucleation of α-synuclein fibrillation, respectively105. In agreement with this inhibitory action of β-synuclein, some positive results of the use of β-synuclein as a means for reducing aggregated α-synuclein levels were obtained in preclinical studies106.
Double-knockout of α- and β-synucleins in mice did not impair basic brain functions or survival, and no significant changes were detected in the ultrastructure of synuclein-deficient synapses, in synaptic plasticity, or in the pool size of synaptic vesicles107. However, these double null animals were characterized by the decreased dopamine levels in their brains and showed selective changes in the levels of synaptic signaling proteins, such as complexins and 14-3-3 proteins107. Recently, analysis of the triple α-synuclein/β-synuclein/γ-synuclein-knockout mice revealed that these proteins have profound effects on the presynaptic architecture and serve as “important orchestrators of presynaptic terminal topography”, regulating presynapse size and organization of the pool of synaptic vesicles108. A systematic analysis of the structural and functional features of the nigrostriatal system in mice with every possible combination of knockout members of the synuclein family revealed that although these proteins have noticeable functional redundancy, some functions are specific for a particular member and therefore the remaining synucleins cannot fully compensate for the deficiency of a lost family member109. For example, β-synuclein was shown to be needed for the efficient maintenance of balance and coordination in aged animals, whereas the presence of α-synuclein is crucial for stabilization of the striatal dopamine level and cannot be compensated by other family members109.
Regulatability angle
Also, a set of recent studies was dedicated to the analysis of various means of regulation of α-synuclein, its normal and pathological functions, and aggregation. It was shown that this protein can be regulated, at the expression level, by the microRNAs miR-34b and miR-34c110 as well as miR-19b, miR-29a, and miR-29c111, by promoter methylation in the α-synuclein gene SNCA112, and by other means of epigenetic-mediated regulation113. Aggregation of α-synuclein can be controlled by antibodies at substoichiometric concentrations (as low as 1:1000 antibody to protein ratio)114. Different monoclonal anti-C-terminal antibodies were shown to differently interact with different forms of α-synuclein (monomeric or aggregated), with the antibodies with the strongest binding to aggregated protein also being the strongest inhibitors of α-synuclein fibrillation and membrane permeabilization115. Aggregation of α-synuclein can be modulated via interaction with the moonlighting 14-3-3 proteins that act as ATP-independent anti-aggregation “holdases”116, by caspase-1-mediated truncation117, by the small secretory chaperone proSAAS118, or by Geum urbanum extract119. The aggregation, toxicity, levels, and secretion of α-synuclein were shown to be controlled by endocytic recycling pathway components, such as Rab8b, Rab11a, Rab13, and Slp5120. Also, interaction with an anti-amyloidogenic agent, ginsenoside Rg1, was shown to inhibit the fibrillation and toxicity of α-synuclein and disaggregate preformed fibrils121. The extracellular α-synuclein can be sorted in extracellular vesicles in a SUMOylation-dependent manner122, whereas the extracellular release of this protein is controlled by the DnaJ/Hsc70 chaperone complexes123. The nuclear accumulation and toxicity of α-synuclein can be regulated by a novel protein, TRIM28124, that controls the fate specification of the neural cells125, whereas loss of lysosomal β-glucocerebrosidase activity promotes global intracellular accumulation and toxicity of α-synuclein126.
Structural polymorphism angle
Protein-chameleon
The immense multifunctionality and related multipathogenicity of α-synuclein suggest that this protein should have a highly amenable structure to be able to do everything ascribed to it. In agreement with this hypothesis, it was recognized early on that α-synuclein is a typical intrinsically disordered protein (IDP) that does not have a stably folded structure under physiological conditions2,127. Furthermore, α-synuclein can serve as an illustrative example of the protein-chameleon concept128, where protein has a highly pliable structure that is extremely sensitive to the environmental conditions. Such a protein is able to morph under the action of numerous factors. As a result, α-synuclein may "stay substantially unfolded, or adopt an amyloidogenic partially folded conformation, or fold into α-helical or β-structural species, both monomeric and oligomeric. Furthermore, it might form several morphologically different types of aggregates, including oligomers (spheres or doughnuts), amorphous aggregates, and or amyloid-like fibrils"128.
The intrinsically disordered nature of α-synuclein is not only a property of the purified protein in vitro but also preserved in vivo, as shown by the analysis of this protein endogenously expressed in the central nervous system, erythrocytes, and mammalian cell lines using native and denaturing gel electrophoresis techniques, size-exclusion chromatography, and an oligomer-specific ELISA129. Compelling support for this idea was also provided by the results of the in-cell NMR analysis of human protein expressed in bacteria130. Very recently, further evidence of the preservation of the disordered nature of monomeric α-synuclein under physiological cell conditions was given by the application of the in-cell NMR and electron paramagnetic resonance spectroscopy to derive the atomic-level information on the structure and dynamics of α-synuclein in different mammalian non-neuronal and neuronal cells131. Based on the molecular dynamics studies, it has also been concluded that a disordered monomer represents the dominant state within the structural ensemble of α-synuclein132.
Residual structure
It was pointed out that although α-synuclein behaves as a highly disordered protein, the solution conformation of this protein is not a random coil but contains some residual structure127,133. This residual structure is extremely sensitive to various environmental factors134. For example, based on the in-cell NMR analysis, it was recently concluded that the conformations of α-synuclein in the cellular environment are more compact than those of the isolated protein in vitro131. These more compact in vivo conformations provide efficient shielding of the residues of the aggregation-prone NAC region, thereby counteracting spontaneous aggregation131 and supporting the “functional misfolding” hypothesis, according to which a polypeptide chain of an IDP can spontaneously misfold “to sequester the preformed elements inside the non-interactive or less-interactive cage, therefore preventing these elements from the unnecessary and unwanted interactions with non-native binding partners”135.
Effects of post-translational modifications
Similar to many other IDPs, α-synuclein is subject to numerous PTMs, such as phosphorylation, ubiquitination, SUMOylation, O-GlcNAcylation, N-terminal acetylation, nitrosylation, and truncation, among which phosphorylation, truncation, and ubiquitination are believed to be the major disease-associated PTMs136,137. Since α-synuclein might have multiple PTMs affecting different residues, and since multiple sites can be subjected to a given PTM (e.g. phosphorylation might occur at S87, S129, or Y125, whereas K12, K21, or K23 can be subjected to ubiquitination), this protein is likely to be highly heterogeneous in its native state. This PTM-driven heterogeneity creates an important problem related to understanding the role of individual PTMs in the structural properties, normal function, misfolding, aggregation, and dysfunction of α-synuclein. Besides the identification of the enzymes involved in regulating these PTMs, a solution to this problem requires the preparation of site-specifically modified proteins. A breakthrough in this direction was achieved in Lashuel’s group, who elaborated a set of enzymatic, synthetic, and semisynthetic strategies for the site-specific and tightly controlled introduction of PTMs at single or multiple sites138–142. These approaches pave the way for generating pure and homogenously modified samples of α-synuclein.
Effects of familial PD mutations
Among the important factors responsible for the modulation of α-synuclein’s residual structure are familial PD mutations. In addition to “traditional” PD mutations (such as A53T3, A30P143, and E46K144) that have been known to the scientific community for a relatively long time, several “new” disease-associated mutations in α-synuclein (H50Q145,146, G51D147,148, and A53E149) were discovered over the past 3 years. These mutations were shown to differently modulate α-synuclein functions and aggregation propensity. For example, the formation of non-fibrillar aggregate (such as oligomers or protofibrils) and not fibrils was accelerated by the A30P mutation150,151. Two other “traditional” PD mutants, A53T and E46K, were both shown to be characterized by accelerated fibrillation150,152–154. Similarly, aggregation and fibrillation of the H50Q mutant were dramatically accelerated155. On the other hand, significant reduction in α-synuclein oligomerization and fibrillation was induced by the G51D and A53E mutations, with the G51D mutant shown to form amorphous aggregates148,156, and with the A53E mutant being able to eventually form very thin amyloid fibrils156–158.
It is known that although mutations can modulate the aggregation propensity of α-synuclein, the global structure of this IDP is rather insensitive to mutations150,153,157,159–163. Furthermore, based on the results of the extensive molecular dynamics simulations in aqueous solution, it has been concluded that fibril structure was not affected by PD-related mutations (A30P, E46K, H50Q, G51D, A53E, and A53T), but the relative stabilities of fibrillar structures and their conformational preferences were altered by mutations164. However, the dynamics and residual structure present in the conformational ensembles of monomeric α-synuclein were clearly affected by the familial PD mutations150,153,157,159–163. For example, by strategic placing of tryptophan residues, the effect of three PD mutations (A53T, E46K, and A30P) on the site-specific structural dynamics of this protein was analyzed165. This analysis showed that mutations affected local conformational flexibility, microenvironment, and solvent exposure165. Analysis of the effects of various mutations on the dynamics of the α-synuclein conformational ensemble revealed that the intramolecular diffusion of this protein is differently affected by aggregation-promoting and aggregation-inhibiting mutations, being correspondingly either drastically slowed down or accelerated in comparison with that of the wild-type protein166. To address the role of tyrosine residues on early stages of α-synuclein aggregation and the effects of these residues on protein dynamics, three modeled Tyr mutants (Y39A, Y133A, and Y125A/Y133A/Y136A) were subjected to all-atom molecular dynamics simulation167. This analysis revealed that the residue Tyr133 plays an important role in driving intramolecular interactions between the hydrophobic residues in the N- and C-terminal regions and is crucial for the fibrillation process167.
Polymorphism of aggregated forms of α-synuclein angle
Not only is monomeric α-synuclein characterized by structural polymorphism but also its oligomeric and insoluble aggregated forms are polymorphic at both structural and functional levels. For example, application of the cryo-electron microscopy image reconstruction technique revealed that oligomers that are kinetically trapped during α-synuclein fibril formation are characterized by different sizes, β-sheet contents, and levels of exposed hydrophobicity168. Also, two morphologically different oligomeric α-synuclein forms were found in human post-mortem PD brain tissue169. The presence of two different types of oligomeric species formed by the wild-type α-synuclein and its familial PD-related mutants A30P, E46K, and A53T was described using hydrogen/deuterium exchange monitored by mass spectrometry170. Although generated at specific conditions, oligomers of the wild-type α-synuclein and its familial PD-related mutants A30P, A53T, E46K, H50Q, and G51D have similar global structures and are composed of a similar number of monomers, but they interact differently with biological membranes171.
Several recent observations are in line with the idea that oligomeric/fibrillar forms of α-synuclein might be functionally different, indicating the presence of a functional polymorphism among the aggregated polymorphs. For example, it was found that fibrillar α-synuclein can trigger inflammatory responses, whereas oligomeric forms of this protein were unable to initiate these cascades172. Although both oligomeric and fibrillar forms of α-synuclein are able to generate free radicals, only the oligomeric protein can reduce endogenous glutathione and cause subsequent neuronal toxicity173. Also, microtubule assembly was shown to be efficiently inhibited by fibrillar α-synuclein, and this fibril-mediated inhibitory effect was greater than that of the oligomeric species (protofibrils)174.
Slight changes in the environmental conditions during the fibrillation process were shown to cause the formation of morphologically different amyloid fibrils with distinctive molecular characteristics that can be inherited by the next generation of fibrils through self-propagation175. Under specific conditions, pure forms of structurally different fibrillar polymorphs of α-synuclein were obtained and characterized structurally and functionally176. In addition to different morphologies, these polymorphs were characterized by different structures, different levels of toxicity, different in vivo and in vitro seeding and propagation properties176, different molecular arrangement in the unit cell, and distinct dynamic features177. Subsequent structural and physical characterization of several fibrillar polymorphs assembled from α-synuclein supported the notion that the same protein can assemble into a variety of large particles with fibrillar shapes, that fibrillar polymorphs are morphologically and nanomechanically different, and that exceptional structural and physical polymorphism is present within the fibrillar form of this protein178. These findings provide mechanical foundation for better understanding of the nature of different α-synuclein strains179 that define the ability of the aggregated α-synuclein to be engaged in different neurodegenerative diseases176.
Strains and transmission angle
It is important to note that the phenomenon of polymorphism of amyloid fibrils is not an exclusive property of α-synuclein but was described for other amyloidogenic proteins180,181 and was linked to the phenomenon of strains originally found in mammalian prion protein182–185 and now described for α-synuclein179, yeast and fungal prions186–188, transthyretin189, and insulin190 as well as Aβ191 and tau protein192. Strain phenomenon is related to the peculiarities of amyloid propagation and transmission, where different aggregated forms of a protein can cause the development of different pathologies. In relation to the subject of this article, the ability of α-synuclein to aggregate into distinct high-molecular-weight assemblies was proposed to be associated with the ability of this protein to cause different synucleinopathies179. This idea is supported by the direct observation of the induction of different synucleinopathies after the administration of the different α-synuclein strains (oligomers, ribbons, and fibrils) by injection into rat brain193.
Another analogy of aggregated α-synuclein to prion protein is its “infectivity”, i.e. the ability to be efficiently transmitted between neurons, thereby supporting the pathological spread within the affected brain during disease progression (e.g. as described by Braak's staging criteria of PD194,195). Although original models describing interneuronal propagation suggested some passive release of aggregated α-synuclein from injured, degenerated, or dead neurons, a recent study showed that intact, relatively healthy neurons are engaged in efficient cell-to-cell passage of α-synuclein196. It was also indicated that endosomal processing might serve as a point of convergence between the transcellular propagation of aggregated α-synuclein and the intracellular trafficking of this protein197. A crucial role of high-affinity binding of preformed α-synuclein fibrils to the lymphocyte-activation gene 3 protein is for the initiation of endocytosis and transmission of aggregated α-synuclein, thereby propagating its toxicity and leading to the loss of dopamine neurons and the development of biochemical and behavioral deficits in vivo198. Importantly, it is not only exogenous fibrillar forms of α-synuclein that are able to promote cellular pathologies, since soluble amyloid oligomers that precede LB formation were also linked to the cell-to-cell transmission of α-synuclein pathology199.
Concluding remarks: proteoforms to the rescue
The knowledge of the peculiar behavior of α-synuclein accumulated over the past two decades provides an illustration of an important notion that the complexity of a biological system is mostly determined by its proteome size and not by the genome size200. In fact, the number of functionally different proteins found in eukaryotic organisms is substantially higher than the number of genes. The functional diversification of proteinaceous products of a gene is achieved by several means, including the allelic variations (i.e. single or multiple point mutations, indels, SNPs) at the DNA level, alternative splicing, and other pre-translational mechanisms affecting mRNA, complemented by a wide spectrum of various PTMs of a polypeptide chain. As a result of all of these events, a set of distinct protein molecules can be created from a single gene, giving rise to the proteoform concept201. Recently, it was proposed that in addition to the aforementioned means that increase the chemical variability of a polypeptide chain, the protein’s structural diversity can be further increased by some other mechanisms, such as intrinsic disorder and function202. In this view, a correlation between protein structure and function is considered a “protein structure–function continuum”, where a given protein exists as a dynamic conformational ensemble containing multiple proteoforms (conformational/basic, inducible/modified, and functioning) characterized by diverse structural features and miscellaneous functions202. In application to α-synuclein, the protein structure–function continuum idea is further enhanced by the presence of remarkable structural and functional variability of its self-aggregated forms. Therefore, due to its ability to oligomerize and aggregate, an intrinsically disordered α-synuclein is characterized by the enormously expanded set of proteoforms where each of the various monomeric, oligomeric, and insoluble species is further split into numerous conformational/basic, inducible/modified, and functioning proteoforms. In other words, as illustrated by Figure 2, consideration of α-synuclein through the proteoform prism provides a logical explanation for its remarkable structural, functional, and dysfunctional multifaceted nature.
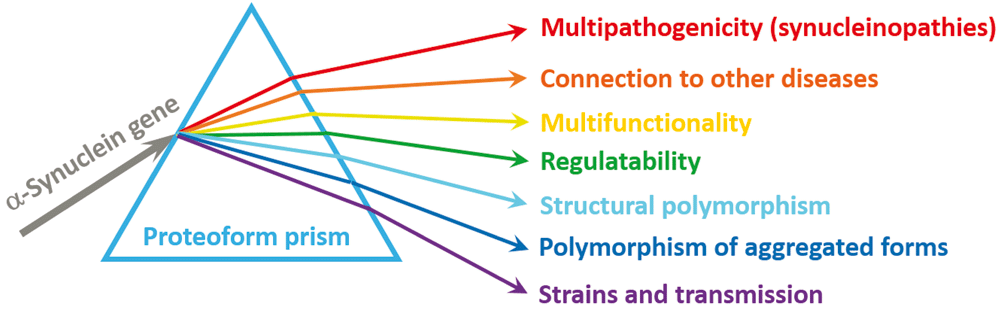
Figure 2. Proteoform concept as a prism for looking at and understanding α-synuclein.
Schematic representation of the proteoform concept using the analogy of a prism. Here, an “incident light” (α-synuclein gene, SNCA), while going through the proteoform “prism” (a set of mechanisms put forward to generate different proteoforms), is “diffracted”, giving rise to a “spectrum” of proteoforms (a set of chemically or structurally different forms of a protein) for conducting different functions.
Outlined in this article, the proteoform-centric view of α-synuclein represents an important shift in understanding the biology and pathology of this protein. In fact, despite the enormous amount of effort expended by the scientific community to find a single most important physiological function of α-synuclein, there is no clear understanding of the unique biological role of this protein as of yet. However, as it follows from a great deal of the published work on α-synuclein, the applicability of the “one sequence – one structure – one function” paradigm to this protein is questionable. In other words, it seems that the search for a single physiological function of α-synuclein may be entirely misplaced, since the intrinsically disordered nature of the protein, which defines the structural polymorphism of the conformational ensemble of its monomeric form, bestows multifunctionality upon α-synuclein. Logical consequences of this structural heterogeneity and multifunctionality of a monomeric protein are the structural polymorphism of aggregated states and a “spectrum of dysfunctions” that define a range of diseases. Therefore, the enormous structural and functional complexity of α-synuclein has serious implications for future studies to understand the biology of this protein and for the development of future therapeutic strategies targeting α-synuclein.
Abbreviations
Aβ, β-amyloid; AD, Alzheimer’s disease; DLB, dementia with Lewy bodies; H3K9, histone H3 lysine-9; IDP, intrinsically disordered protein; LB, Lewy body; LN, Lewy neurite; MSA, multiple system atrophy; NAC, non-amyloid-β component; PD, Parkinson’s disease; PP2A, protein phosphatase-2A; PTM, post-translational modification.
Competing interests
The author declares that he has no competing interests.
Grant information
The author(s) declared that no grants were involved in supporting this work.
Faculty Opinions recommendedReferences
- 1.
Maroteaux L, Scheller RH:
The rat brain synucleins; family of proteins transiently associated with neuronal membrane.
Brain Res Mol Brain Res.
1991; 11(3–4): 335–43. PubMed Abstract
| Publisher Full Text
- 2.
Weinreb PH, Zhen W, Poon AW, et al.:
NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded.
Biochemistry.
1996; 35(43): 13709–15. PubMed Abstract
| Publisher Full Text
- 3.
Polymeropoulos MH, Lavedan C, Leroy E, et al.:
Mutation in the alpha-synuclein gene identified in families with Parkinson's disease.
Science.
1997; 276(5321): 2045–7. PubMed Abstract
| Publisher Full Text
- 4.
Spillantini MG, Schmidt ML, Lee VM, et al.:
Alpha-synuclein in Lewy bodies.
Nature.
1997; 388(6645): 839–40. PubMed Abstract
| Publisher Full Text
- 5.
Lewy FH:
Paralysis Agitans. Pathologische Anatomie. In: Lewandowski M (ed), Handbuch der Neurologie. Springer, Berlin, 1912; 920–933.
- 6.
Forno LS:
Neuropathology of Parkinson's disease.
J Neuropathol Exp Neurol.
1996; 55(3): 259–72. PubMed Abstract
| Publisher Full Text
- 7.
McKeith IG, Dickson DW, Lowe J, et al.:
Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium.
Neurology.
2005; 65(12): 1863–72. PubMed Abstract
| Publisher Full Text
- 8.
Wakabayashi K, Yoshimoto M, Tsuji S, et al.:
Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy.
Neurosci Lett.
1998; 249(2–3): 180–2. PubMed Abstract
| Publisher Full Text
- 9.
Spillantini MG, Crowther RA, Jakes R, et al.:
Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson's disease and dementia with Lewy bodies.
Neurosci Lett.
1998; 251(3): 205–8. PubMed Abstract
| Publisher Full Text
- 10.
Gai WP, Power JH, Blumbergs PC, et al.:
Multiple-system atrophy: a new alpha-synuclein disease?
Lancet.
1998; 352(9127): 547–8. PubMed Abstract
| Publisher Full Text
- 11.
Trojanowski JQ, Goedert M, Iwatsubo T, et al.:
Fatal attractions: abnormal protein aggregation and neuron death in Parkinson's disease and Lewy body dementia.
Cell Death Differ.
1998; 5(10): 832–7. PubMed Abstract
| Publisher Full Text
- 12.
Takeda A, Mallory M, Sundsmo M, et al.:
Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders.
Am J Pathol.
1998; 152(2): 367–72. PubMed Abstract
| Free Full Text
- 13.
Lücking CB, Brice A:
Alpha-synuclein and Parkinson's disease.
Cell Mol Life Sci.
2000; 57(13–14): 1894–908. PubMed Abstract
| Publisher Full Text
- 14.
Arawaka S, Saito Y, Murayama S, et al.:
Lewy body in neurodegeneration with brain iron accumulation type 1 is immunoreactive for alpha-synuclein.
Neurology.
1998; 51(3): 887–9. PubMed Abstract
| Publisher Full Text
- 15.
Wakabayashi K, Matsumoto K, Takayama K, et al.:
NACP, a presynaptic protein, immunoreactivity in Lewy bodies in Parkinson's disease.
Neurosci Lett.
1997; 239(1): 45–8. PubMed Abstract
| Publisher Full Text
- 16.
Spillantini MG, Crowther RA, Jakes R, et al.:
alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies.
Proc Natl Acad Sci U S A.
1998; 95(11): 6469–73. PubMed Abstract
| Free Full Text
- 17.
Spillantini MG, Goedert M:
The alpha-synucleinopathies: Parkinson's disease, dementia with Lewy bodies, and multiple system atrophy.
Ann N Y Acad Sci.
2000; 920: 16–27. PubMed Abstract
| Publisher Full Text
- 18.
Goedert M:
Filamentous nerve cell inclusions in neurodegenerative diseases: tauopathies and alpha-synucleinopathies.
Philos Trans R Soc Lond B Biol Sci.
1999; 354(1386): 1101–18. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 19.
Galvin JE, Lee VM, Trojanowski JQ:
Synucleinopathies: clinical and pathological implications.
Arch Neurol.
2001; 58(2): 186–90. PubMed Abstract
| Publisher Full Text
- 20.
Trojanowski JQ, Lee VM:
Parkinson's disease and related alpha-synucleinopathies are brain amyloidoses.
Ann N Y Acad Sci.
2003; 991: 107–10. PubMed Abstract
| Publisher Full Text
- 21.
Lundvig D, Lindersson E, Jensen PH:
Pathogenic effects of alpha-synuclein aggregation.
Brain Res Mol Brain Res.
2005; 134(1): 3–17. PubMed Abstract
| Publisher Full Text
- 22.
den Hartog Jager WA, Bethlem J:
The distribution of Lewy bodies in the central and autonomic nervous systems in idiopathic paralysis agitans.
J Neurol Neurosurg Psychiatry.
1960; 23(4): 283–90. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 23.
Kosaka K:
Lewy bodies in cerebral cortex, report of three cases.
Acta Neuropathol.
1978; 42(2): 127–34. PubMed Abstract
| Publisher Full Text
- 24.
Kosaka K, Mehraein P:
Dementia-Parkinsonism syndrome with numerous Lewy bodies and senile plaques in cerebral cortex.
Arch Psychiatr Nervenkr (1970).
1979; 226(4): 241–50. PubMed Abstract
| Publisher Full Text
- 25.
Seidel K, Mahlke J, Siswanto S, et al.:
The brainstem pathologies of Parkinson's disease and dementia with Lewy bodies.
Brain Pathol.
2015; 25(2): 121–35. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 26.
Roberts RF, Wade-Martins R, Alegre-Abarrategui J:
Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinson's disease brain.
Brain.
2015; 138(Pt 6): 1642–57. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 27.
Iwai A, Masliah E, Yoshimoto M, et al.:
The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system.
Neuron.
1995; 14(2): 467–75. PubMed Abstract
| Publisher Full Text
- 28.
Poças GM, Branco-Santos J, Herrera F, et al.:
α-Synuclein modifies mutant huntingtin aggregation and neurotoxicity in Drosophila.
Hum Mol Genet.
2015; 24(7): 1898–907. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 29.
Sengupta U, Guerrero-Muñoz MJ, Castillo-Carranza DL, et al.:
Pathological interface between oligomeric alpha-synuclein and tau in synucleinopathies.
Biol Psychiatry.
2015; 78(10): 672–83. PubMed Abstract
| Publisher Full Text
- 30.
Choi BK, Kim JY, Cha MY, et al.:
β-Amyloid and α-synuclein cooperate to block SNARE-dependent vesicle fusion.
Biochemistry.
2015; 54(9): 1831–40. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 31.
Bachhuber T, Katzmarski N, McCarter JF, et al.:
Inhibition of amyloid-β plaque formation by α-synuclein.
Nat Med.
2015; 21(7): 802–7. PubMed Abstract
| Publisher Full Text
- 32.
Miki Y, Tanji K, Mori F, et al.:
AMBRA1, a novel α-synuclein-binding protein, is implicated in the pathogenesis of multiple system atrophy.
Brain Pathol.
2016. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 33.
Beadle GW, Tatum EL:
Genetic Control of Biochemical Reactions in Neurospora.
Proc Natl Acad Sci U S A.
1941; 27(11): 499–506. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 34.
Dev KK, Hofele K, Barbieri S, et al.:
Part II: alpha-synuclein and its molecular pathophysiological role in neurodegenerative disease.
Neuropharmacology.
2003; 45(1): 14–44. PubMed Abstract
| Publisher Full Text
- 35.
Ottolini D, Calí T, Szabò I, et al.:
Alpha-synuclein at the intracellular and the extracellular side: functional and dysfunctional implications.
Biol Chem.
2017; 398(1): 77–100. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 36.
Uversky VN, Li J, Fink AL:
Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson's disease and heavy metal exposure.
J Biol Chem.
2001; 276(47): 44284–96. PubMed Abstract
| Publisher Full Text
- 37.
Santner A, Uversky VN:
Metalloproteomics and metal toxicology of α-synuclein.
Metallomics.
2010; 2(6): 378–92. PubMed Abstract
| Publisher Full Text
- 38.
Ahmad A, Burns CS, Fink AL, et al.:
Peculiarities of copper binding to alpha-synuclein.
J Biomol Struct Dyn.
2012; 29(4): 825–42. PubMed Abstract
| Publisher Full Text
- 39.
Carboni E, Lingor P:
Insights on the interaction of alpha-synuclein and metals in the pathophysiology of Parkinson's disease.
Metallomics.
2015; 7(3): 395–404. PubMed Abstract
| Publisher Full Text
- 40.
Uversky VN, Li J, Bower K, et al.:
Synergistic effects of pesticides and metals on the fibrillation of alpha-synuclein: implications for Parkinson's disease.
Neurotoxicology.
2002; 23(4–5): 527–36. PubMed Abstract
| Publisher Full Text
- 41.
Uversky VN, Li J, Fink AL:
Pesticides directly accelerate the rate of alpha-synuclein fibril formation: a possible factor in Parkinson's disease.
FEBS Lett.
2001; 500(3): 105–8. PubMed Abstract
| Publisher Full Text
- 42.
Maturana MG, Pinheiro AS, de Souza TL, et al.:
Unveiling the role of the pesticides paraquat and rotenone on α-synuclein fibrillation in vitro.
Neurotoxicology.
2015; 46: 35–43. PubMed Abstract
| Publisher Full Text
- 43.
Emanuele M, Chieregatti E:
Mechanisms of alpha-synuclein action on neurotransmission: cell-autonomous and non-cell autonomous role.
Biomolecules.
2015; 5(2): 865–92. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 44.
da Costa CA, Ancolio K, Checler F:
Wild-type but not Parkinson's disease-related ala-53 --> Thr mutant alpha -synuclein protects neuronal cells from apoptotic stimuli.
J Biol Chem.
2000; 275(31): 24065–9. PubMed Abstract
| Publisher Full Text
- 45.
Uversky VN:
Alpha-synuclein misfolding and neurodegenerative diseases.
Curr Protein Pept Sci.
2008; 9(5): 507–40. PubMed Abstract
| Publisher Full Text
- 46.
Payton JE, Perrin RJ, Clayton DF, et al.:
Protein-protein interactions of alpha-synuclein in brain homogenates and transfected cells.
Brain Res Mol Brain Res.
2001; 95(1–2): 138–45. PubMed Abstract
| Publisher Full Text
- 47.
Jin J, Li GJ, Davis J, et al.:
Identification of novel proteins associated with both alpha-synuclein and DJ-1.
Mol Cell Proteomics.
2007; 6(5): 845–59. PubMed Abstract
| Publisher Full Text
- 48.
Abeliovich A, Schmitz Y, Fariñas I, et al.:
Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system.
Neuron.
2000; 25(1): 239–52. PubMed Abstract
| Publisher Full Text
- 49.
Cabin DE, Shimazu K, Murphy D, et al.:
Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein.
J Neurosci.
2002; 22(20): 8797–807. PubMed Abstract
- 50.
Yavich L, Jäkälä P, Tanila H:
Abnormal compartmentalization of norepinephrine in mouse dentate gyrus in alpha-synuclein knockout and A30P transgenic mice.
J Neurochem.
2006; 99(3): 724–32. PubMed Abstract
| Publisher Full Text
- 51.
Oksman M, Tanila H, Yavich L:
Brain reward in the absence of alpha-synuclein.
Neuroreport.
2006; 17(11): 1191–4. PubMed Abstract
| Publisher Full Text
- 52.
Ettle B, Kuhbandner K, Jörg S, et al.:
α-Synuclein deficiency promotes neuroinflammation by increasing Th1 cell-mediated immune responses.
J Neuroinflammation.
2016; 13(1): 201. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 53.
Dauer W, Kholodilov N, Vila M, et al.:
Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP.
Proc Natl Acad Sci U S A.
2002; 99(22): 14524–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 54.
Robertson DC, Schmidt O, Ninkina N, et al.:
Developmental loss and resistance to MPTP toxicity of dopaminergic neurones in substantia nigra pars compacta of gamma-synuclein, alpha-synuclein and double alpha/gamma-synuclein null mutant mice.
J Neurochem.
2004; 89(5): 1126–36. PubMed Abstract
| Publisher Full Text
- 55.
Drolet RE, Behrouz B, Lookingland KJ, et al.:
Mice lacking alpha-synuclein have an attenuated loss of striatal dopamine following prolonged chronic MPTP administration.
Neurotoxicology.
2004; 25(5): 761–9. PubMed Abstract
| Publisher Full Text
- 56.
Golovko MY, Faergeman NJ, Cole NB, et al.:
Alpha-synuclein gene deletion decreases brain palmitate uptake and alters the palmitate metabolism in the absence of alpha-synuclein palmitate binding.
Biochemistry.
2005; 44(23): 8251–9. PubMed Abstract
| Publisher Full Text
- 57.
Golovko MY, Rosenberger TA, Feddersen S, et al.:
Alpha-synuclein gene ablation increases docosahexaenoic acid incorporation and turnover in brain phospholipids.
J Neurochem.
2007; 101(1): 201–11. PubMed Abstract
| Publisher Full Text
- 58.
Golovko MY, Rosenberger TA, Faergeman NJ, et al.:
Acyl-CoA synthetase activity links wild-type but not mutant alpha-synuclein to brain arachidonate metabolism.
Biochemistry.
2006; 45(22): 6956–66. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 59.
Barceló-Coblijn G, Golovko MY, Weinhofer I, et al.:
Brain neutral lipids mass is increased in alpha-synuclein gene-ablated mice.
J Neurochem.
2007; 101(1): 132–41. PubMed Abstract
| Publisher Full Text
- 60.
Kermer P, Köhn A, Schnieder M, et al.:
BAG1 is neuroprotective in in vivo and in vitro models of Parkinson's disease.
J Mol Neurosci.
2015; 55(3): 587–95. PubMed Abstract
| Publisher Full Text
- 61.
Savolainen MH, Yan X, Myöhänen TT, et al.:
Prolyl oligopeptidase enhances α-synuclein dimerization via direct protein-protein interaction.
J Biol Chem.
2015; 290(8): 5117–26. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 62.
Betzer C, Movius AJ, Shi M, et al.:
Identification of synaptosomal proteins binding to monomeric and oligomeric α-synuclein.
PLoS One.
2015; 10(2): e0116473. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 63.
Guardia-Laguarta C, Area-Gomez E, Schon EA, et al.:
Novel subcellular localization for α-synuclein: possible functional consequences.
Front Neuroanat.
2015; 9: 17. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 64.
Chai YJ, Sierecki E, Tomatis VM, et al.:
Munc18-1 is a molecular chaperone for α-synuclein, controlling its self-replicating aggregation.
J Cell Biol.
2016; 214(6): 705–18. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 65.
Zaltieri M, Grigoletto J, Longhena F, et al.:
α-synuclein and synapsin III cooperatively regulate synaptic function in dopamine neurons.
J Cell Sci.
2015; 128(13): 2231–43. PubMed Abstract
| Publisher Full Text
- 66.
Rostovtseva TK, Gurnev PA, Protchenko O, et al.:
α-Synuclein Shows High Affinity Interaction with Voltage-dependent Anion Channel, Suggesting Mechanisms of Mitochondrial Regulation and Toxicity in Parkinson Disease.
J Biol Chem.
2015; 290(30): 18467–77. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 67.
Kakish J, Tavassoly O, Lee JS:
Rasagiline, a suicide inhibitor of monoamine oxidases, binds reversibly to α-synuclein.
ACS Chem Neurosci.
2015; 6(2): 347–55. PubMed Abstract
| Publisher Full Text
- 68.
Jinsmaa Y, Cooney A, Sullivan P, et al.:
The serotonin aldehyde, 5-HIAL, oligomerizes alpha-synuclein.
Neurosci Lett.
2015; 590: 134–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 69.
Schapansky J, Nardozzi JD, LaVoie MJ:
The complex relationships between microglia, alpha-synuclein, and LRRK2 in Parkinson's disease.
Neuroscience.
2015; 302: 74–88. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 70.
Zhang H, Duan C, Yang H:
Defective autophagy in Parkinson's disease: lessons from genetics.
Mol Neurobiol.
2015; 51(1): 89–104. PubMed Abstract
| Publisher Full Text
- 71.
Mishra AK, ur Rasheed MS, Shukla S, et al.:
Aberrant autophagy and parkinsonism: does correction rescue from disease progression?
Mol Neurobiol.
2015; 51(3): 893–908. PubMed Abstract
| Publisher Full Text
- 72.
Allen Reish HE, Standaert DG:
Role of α-synuclein in inducing innate and adaptive immunity in Parkinson disease.
J Parkinsons Dis.
2015; 5(1): 1–19. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 73.
Shi Z, Sachs JN, Rhoades E, et al.:
Biophysics of α-synuclein induced membrane remodelling.
Phys Chem Chem Phys.
2015; 17(24): 15561–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 74.
Fakhree MA, Zijlstra N, Raiss CC, et al.:
The number of α-synuclein proteins per vesicle gives insights into its physiological function.
Sci Rep.
2016; 6: 30658. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 75.
Breda C, Nugent ML, Estranero JG, et al.:
Rab11 modulates α-synuclein-mediated defects in synaptic transmission and behaviour.
Hum Mol Genet.
2015; 24(4): 1077–91. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 76.
Volpicelli-Daley LA:
Effects of α-synuclein on axonal transport.
Neurobiol Dis.
2016; pii: S0969-9961(16)30289-3. PubMed Abstract
| Publisher Full Text
- 77.
Norris KL, Hao R, Chen L, et al.:
Convergence of Parkin, PINK1, and α-Synuclein on Stress-induced Mitochondrial Morphological Remodeling.
J Biol Chem.
2015; 290(22): 13862–74. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 78.
Guardia-Laguarta C, Area-Gomez E, Schon EA, et al.:
A new role for α-synuclein in Parkinson's disease: Alteration of ER-mitochondrial communication.
Mov Disord.
2015; 30(8): 1026–33. PubMed Abstract
| Publisher Full Text
- 79.
Cartelli D, Aliverti A, Barbiroli A, et al.:
alpha-Synuclein is a Novel Microtubule Dynamase.
Sci Rep.
2016; 6: 33289. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 80.
Credle JJ, Forcelli PA, Delannoy M, et al.:
alpha-Synuclein-mediated inhibition of ATF6 processing into COPII vesicles disrupts UPR signaling in Parkinson's disease.
Neurobiol Dis.
2015; 76: 112–25. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 81.
Park SC, Moon JC, Shin SY, et al.:
Functional characterization of alpha-synuclein protein with antimicrobial activity.
Biochem Biophys Res Commun.
2016; 478(2): 924–8. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 82.
Kumar A, Leinisch F, Kadiiska MB, et al.:
Formation and Implications of Alpha-Synuclein Radical in Maneb- and Paraquat-Induced Models of Parkinson's Disease.
Mol Neurobiol.
2016; 53(5): 2983–94. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 83.
Gispert S, Kurz A, Brehm N, et al.:
Complexin-1 and Foxp1 Expression Changes Are Novel Brain Effects of Alpha-Synuclein Pathology.
Mol Neurobiol.
2015; 52(1): 57–63. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 84.
Sugeno N, Jäckel S, Voigt A, et al.:
α-Synuclein enhances histone H3 lysine-9 dimethylation and H3K9me2-dependent transcriptional responses.
Sci Rep.
2016; 6: 36328. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 85.
Liang W, Miao S, Zhang B, et al.:
Synuclein γ protects Akt and mTOR and renders tumor resistance to Hsp90 disruption.
Oncogene.
2015; 34(18): 2398–405. PubMed Abstract
| Publisher Full Text
- 86.
Hua G, Xiaolei L, Weiwei Y, et al.:
Protein phosphatase 2A is involved in the tyrosine hydroxylase phosphorylation regulated by α-synuclein.
Neurochem Res.
2015; 40(3): 428–37. PubMed Abstract
| Publisher Full Text
- 87.
Navarria L, Zaltieri M, Longhena F, et al.:
Alpha-synuclein modulates NR2B-containing NMDA receptors and decreases their levels after rotenone exposure.
Neurochem Int.
2015; 85–86: 14–23. PubMed Abstract
| Publisher Full Text
- 88.
Baek HJ, Yoon JH, Ann EJ, et al.:
Alpha-synuclein negatively regulates Notch1 intracellular domain protein stability through promoting interaction with Fbw7.
Neurosci Lett.
2015; 600: 6–11. PubMed Abstract
| Publisher Full Text
- 89.
Clayton DF, George JM:
Synucleins in synaptic plasticity and neurodegenerative disorders.
J Neurosci Res.
1999; 58(1): 120–9. PubMed Abstract
| Publisher Full Text
- 90.
Uéda K, Fukushima H, Masliah E, et al.:
Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease.
Proc Natl Acad Sci U S A.
1993; 90(23): 11282–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 91.
Maroteaux L, Campanelli JT, Scheller RH:
Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal.
J Neurosci.
1988; 8(8): 2804–15. PubMed Abstract
- 92.
Jakes R, Spillantini MG, Goedert M:
Identification of two distinct synucleins from human brain.
FEBS Lett.
1994; 345(1): 27–32. PubMed Abstract
| Publisher Full Text
- 93.
Nakajo S, Tsukada K, Omata K, et al.:
A new brain-specific 14-kDa protein is a phosphoprotein. Its complete amino acid sequence and evidence for phosphorylation.
Eur J Biochem.
1993; 217(3): 1057–63. PubMed Abstract
| Publisher Full Text
- 94.
Tobe T, Nakajo S, Tanaka A, et al.:
Cloning and characterization of the cDNA encoding a novel brain-specific 14-kDa protein.
J Neurochem.
1992; 59(5): 1624–9. PubMed Abstract
| Publisher Full Text
- 95.
Ji H, Liu YE, Jia T, et al.:
Identification of a breast cancer-specific gene, BCSG1, by direct differential cDNA sequencing.
Cancer Res.
1997; 57(4): 759–64. PubMed Abstract
- 96.
Ninkina N, Alimova-Kost MV, Paterson JW, et al.:
Organization, expression and polymorphism of the human persyn gene.
Hum Mol Genet.
1998; 7(9): 1417–24. PubMed Abstract
| Publisher Full Text
- 97.
Buchman VL, Hunter HJ, Pinõn LG, et al.:
Persyn, a member of the synuclein family, has a distinct pattern of expression in the developing nervous system.
J Neurosci.
1998; 18(22): 9335–41. PubMed Abstract
- 98.
Lavedan C, Leroy E, Dehejia A, et al.:
Identification, localization and characterization of the human gamma-synuclein gene.
Hum Genet.
1998; 103(1): 106–12. PubMed Abstract
| Publisher Full Text
- 99.
Clayton DF, George JM:
The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease.
Trends Neurosci.
1998; 21(6): 249–54. PubMed Abstract
| Publisher Full Text
- 100.
Galvin JE, Uryu K, Lee VM, et al.:
Axon pathology in Parkinson's disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein.
Proc Natl Acad Sci U S A.
1999; 96(23): 13450–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 101.
Uversky VN, Li J, Souillac P, et al.:
Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of alpha-synuclein assembly by beta- and gamma-synucleins.
J Biol Chem.
2002; 277(14): 11970–8. PubMed Abstract
| Publisher Full Text
- 102.
Yamin G, Munishkina LA, Karymov MA, et al.:
Forcing nonamyloidogenic beta-synuclein to fibrillate.
Biochemistry.
2005; 44(25): 9096–107. PubMed Abstract
| Publisher Full Text
- 103.
Hashimoto M, Rockenstein E, Mante M, et al.:
beta-Synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor.
Neuron.
2001; 32(2): 213–23. PubMed Abstract
| Publisher Full Text
- 104.
Janowska MK, Wu KP, Baum J:
Unveiling transient protein-protein interactions that modulate inhibition of alpha-synuclein aggregation by beta-synuclein, a pre-synaptic protein that co-localizes with alpha-synuclein.
Sci Rep.
2015; 5: 15164. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 105.
Brown JW, Buell AK, Michaels TC, et al.:
β-Synuclein suppresses both the initiation and amplification steps of α-synuclein aggregation via competitive binding to surfaces.
Sci Rep.
2016; 6: 36010. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 106.
Tolmasov M, Djaldetti R, Lev N, et al.:
Pathological and clinical aspects of alpha/beta synuclein in Parkinson's disease and related disorders.
Expert Rev Neurother.
2016; 16(5): 505–13. PubMed Abstract
| Publisher Full Text
- 107.
Chandra S, Fornai F, Kwon H, et al.:
Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions.
Proc Natl Acad Sci U S A.
2004; 101(41): 14966–71. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 108.
Vargas KJ, Schrod N, Davis T, et al.:
Synucleins Have Multiple Effects on Presynaptic Architecture.
Cell Rep.
2017; 18(1): 161–73. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 109.
Connor-Robson N, Peters OM, Millership S, et al.:
Combinational losses of synucleins reveal their differential requirements for compensating age-dependent alterations in motor behavior and dopamine metabolism.
Neurobiol Aging.
2016; 46: 107–12. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 110.
Kabaria S, Choi DC, Chaudhuri AD, et al.:
Inhibition of miR-34b and miR-34c enhances α-synuclein expression in Parkinson's disease.
FEBS Lett.
2015; 589(3): 319–25. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 111.
Fernández-Santiago R, Iranzo A, Gaig C, et al.:
MicroRNA association with synucleinopathy conversion in rapid eye movement behavior disorder.
Ann Neurol.
2015; 77(5): 895–901. PubMed Abstract
| Publisher Full Text
- 112.
Pihlstrøm L, Berge V, Rengmark A, et al.:
Parkinson's disease correlates with promoter methylation in the α-synuclein gene.
Mov Disord.
2015; 30(4): 577–80. PubMed Abstract
| Publisher Full Text
- 113.
Pavlou MA, Pinho R, Paiva I, et al.:
The yin and yang of α-synuclein-associated epigenetics in Parkinson's disease.
Brain.
2016; pii: aww227. PubMed Abstract
| Publisher Full Text
- 114.
Breydo L, Morgan D, Uversky VN:
Pseudocatalytic Antiaggregation Activity of Antibodies: Immunoglobulins can Influence α-Synuclein Aggregation at Substoichiometric Concentrations.
Mol Neurobiol.
2016; 53(3): 1949–58. PubMed Abstract
| Publisher Full Text
- 115.
Sahin C, Lorenzen N, Lemminger L, et al.:
Antibodies against the C-terminus of α-synuclein modulate its fibrillation.
Biophys Chem.
2017; 220: 34–41. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 116.
Sluchanko NN, Gusev NB:
Moonlighting chaperone-like activity of the universal regulatory 14-3-3 proteins.
FEBS J.
2016. PubMed Abstract
| Publisher Full Text
- 117.
Wang W, Nguyen LT, Burlak C, et al.:
Caspase-1 causes truncation and aggregation of the Parkinson's disease-associated protein α-synuclein.
Proc Natl Acad Sci U S A.
2016; 113(34): 9587–92. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 118.
Jarvela TS, Lam HA, Helwig M, et al.:
The neural chaperone proSAAS blocks α-synuclein fibrillation and neurotoxicity.
Proc Natl Acad Sci U S A.
2016; 113(32): E4708–15. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 119.
Lobbens ES, Breydo L, Skamris T, et al.:
Mechanistic study of the inhibitory activity of Geum urbanum extract against α-Synuclein fibrillation.
Biochim Biophys Acta.
2016; 1864(9): 1160–9. PubMed Abstract
| Publisher Full Text
- 120.
Gonçalves SA, Macedo D, Raquel H, et al.:
shRNA-Based Screen Identifies Endocytic Recycling Pathway Components That Act as Genetic Modifiers of Alpha-Synuclein Aggregation, Secretion and Toxicity.
PLoS Genet.
2016; 12(4): e1005995. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 121.
Ardah MT, Paleologou KE, Lv G, et al.:
Ginsenoside Rb1 inhibits fibrillation and toxicity of alpha-synuclein and disaggregates preformed fibrils.
Neurobiol Dis.
2015; 74: 89–101. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 122.
Kunadt M, Eckermann K, Stuendl A, et al.:
Extracellular vesicle sorting of α-Synuclein is regulated by sumoylation.
Acta Neuropathol.
2015; 129(5): 695–713. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 123.
Fontaine SN, Zheng D, Sabbagh JJ, et al.:
DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins.
EMBO J.
2016; 35(14): 1537–49. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 124.
Rousseaux MW, de Haro M, Lasagna-Reeves CA, et al.:
TRIM28 regulates the nuclear accumulation and toxicity of both alpha-synuclein and tau.
eLife.
2016; 5: pii: e19809. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 125.
Pavlou MA, Colombo N, Fuertes-Alvarez S, et al.:
Expression of the Parkinson's Disease-Associated Gene Alpha-Synuclein is Regulated by the Neuronal Cell Fate Determinant TRIM32.
Mol Neurobiol.
2016; 1–14. PubMed Abstract
| Publisher Full Text
- 126.
Wong YC, Krainc D:
Lysosomal trafficking defects link Parkinson's disease with Gaucher's disease.
Mov Disord.
2016; 31(11): 1610–8. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 127.
Uversky VN, Li J, Fink AL:
Evidence for a partially folded intermediate in alpha-synuclein fibril formation.
J Biol Chem.
2001; 276(14): 10737–44. PubMed Abstract
| Publisher Full Text
- 128.
Uversky VN:
A protein-chameleon: conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders.
J Biomol Struct Dyn.
2003; 21(2): 211–34. PubMed Abstract
| Publisher Full Text
- 129.
Fauvet B, Mbefo MK, Fares M, et al.:
α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer.
J Biol Chem.
2012; 287(19): 15345–64. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 130.
Binolfi A, Theillet FX, Selenko P:
Bacterial in-cell NMR of human α-synuclein: a disordered monomer by nature?
Biochem Soc Trans.
2012; 40(5): 950–4. PubMed Abstract
| Publisher Full Text
- 131.
Theillet FX, Binolfi A, Bekei B, et al.:
Structural disorder of monomeric α-synuclein persists in mammalian cells.
Nature.
2016; 530(7588): 45–50. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 132.
Gurry T, Ullman O, Fisher CK, et al.:
The dynamic structure of α-synuclein multimers.
J Am Chem Soc.
2013; 135(10): 3865–72. PubMed Abstract
| Publisher Full Text
- 133.
Eliezer D, Kutluay E, Bussell R Jr, et al.:
Conformational properties of alpha-synuclein in its free and lipid-associated states.
J Mol Biol.
2001; 307(4): 1061–73. PubMed Abstract
| Publisher Full Text
- 134.
Uversky VN, Eliezer D:
Biophysics of Parkinson's disease: structure and aggregation of alpha-synuclein.
Curr Protein Pept Sci.
2009; 10(5): 483–99. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 135.
Uversky VN:
Intrinsically disordered proteins may escape unwanted interactions via functional misfolding.
Biochim Biophys Acta.
2011; 1814(5): 693–712. PubMed Abstract
| Publisher Full Text
- 136.
Oueslati A, Fournier M, Lashuel HA:
Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein: implications for Parkinson's disease pathogenesis and therapies.
Prog Brain Res.
2010; 183: 115–45. PubMed Abstract
| Publisher Full Text
- 137.
Schmid AW, Fauvet B, Moniatte M, et al.:
Alpha-synuclein post-translational modifications as potential biomarkers for Parkinson disease and other synucleinopathies.
Mol Cell Proteomics.
2013; 12(12): 3543–58. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 138.
Hejjaoui M, Haj-Yahya M, Kumar KS, et al.:
Towards elucidation of the role of ubiquitination in the pathogenesis of Parkinson's disease with semisynthetic ubiquitinated α-synuclein.
Angew Chem Int Ed Engl.
2011; 50(2): 405–9. PubMed Abstract
| Publisher Full Text
- 139.
Hejjaoui M, Butterfield S, Fauvet B, et al.:
Elucidating the role of C-terminal post-translational modifications using protein semisynthesis strategies: α-synuclein phosphorylation at tyrosine 125.
J Am Chem Soc.
2012; 134(11): 5196–210. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 140.
Haj-Yahya M, Fauvet B, Herman-Bachinsky Y, et al.:
Synthetic polyubiquitinated α-Synuclein reveals important insights into the roles of the ubiquitin chain in regulating its pathophysiology.
Proc Natl Acad Sci U S A.
2013; 110(44): 17726–31. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 141.
Burai R, Ait-Bouziad N, Chiki A, et al.:
Elucidating the Role of Site-Specific Nitration of α-Synuclein in the Pathogenesis of Parkinson's Disease via Protein Semisynthesis and Mutagenesis.
J Am Chem Soc.
2015; 137(15): 5041–52. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 142.
Fauvet B, Lashuel HA:
Semisynthesis and Enzymatic Preparation of Post-translationally Modified α-Synuclein.
Methods Mol Biol.
2016; 1345: 3–20. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 143.
Kruger R, Kuhn W, Müller T, et al.:
Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease.
Nat Genet.
1998; 18(2): 106–8. PubMed Abstract
| Publisher Full Text
- 144.
Zarranz JJ, Alegre J, Gómez-Esteban JC, et al.:
The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia.
Ann Neurol.
2004; 55(2): 164–73. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 145.
Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, et al.:
Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease.
Mov Disord.
2013; 28(6): 811–3. PubMed Abstract
| Publisher Full Text
- 146.
Proukakis C, Dudzik CG, Brier T, et al.:
A novel α-synuclein missense mutation in Parkinson disease.
Neurology.
2013; 80(11): 1062–4. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 147.
Kiely AP, Asi YT, Kara E, et al.:
α-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson's disease and multiple system atrophy?
Acta Neuropathol.
2013; 125(5): 753–69. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 148.
Lesage S, Anheim M, Letournel F, et al.:
G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome.
Ann Neurol.
2013; 73(4): 459–71. PubMed Abstract
| Publisher Full Text
- 149.
Pasanen P, Myllykangas L, Siitonen M, et al.:
Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson's disease-type pathology.
Neurobiol Aging.
2014; 35(9): 2180.e1–5. PubMed Abstract
| Publisher Full Text
- 150.
Li J, Uversky VN, Fink AL:
Effect of familial Parkinson's disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human alpha-synuclein.
Biochemistry.
2001; 40(38): 11604–13. PubMed Abstract
| Publisher Full Text
- 151.
Lemkau LR, Comellas G, Kloepper KD, et al.:
Mutant protein A30P α-synuclein adopts wild-type fibril structure, despite slower fibrillation kinetics.
J Biol Chem.
2012; 287(14): 11526–32. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 152.
Conway KA, Harper JD, Lansbury PT:
Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease.
Nat Med.
1998; 4(11): 1318–20. PubMed Abstract
| Publisher Full Text
- 153.
Fredenburg RA, Rospigliosi C, Meray RK, et al.:
The impact of the E46K mutation on the properties of alpha-synuclein in its monomeric and oligomeric states.
Biochemistry.
2007; 46(24): 7107–18. PubMed Abstract
| Publisher Full Text
- 154.
Pandey N, Schmidt RE, Galvin JE:
The alpha-synuclein mutation E46K promotes aggregation in cultured cells.
Exp Neurol.
2006; 197(2): 515–20. PubMed Abstract
| Publisher Full Text
- 155.
Khalaf O, Fauvet B, Oueslati A, et al.:
The H50Q mutation enhances α-synuclein aggregation, secretion, and toxicity.
J Biol Chem.
2014; 289(32): 21856–76. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 156.
Rutherford NJ, Moore BD, Golde TE, et al.:
Divergent effects of the H50Q and G51D SNCA mutations on the aggregation of α-synuclein.
J Neurochem.
2014; 131(6): 859–67. PubMed Abstract
| Publisher Full Text
- 157.
Ghosh D, Sahay S, Ranjan P, et al.:
The newly discovered Parkinson's disease associated Finnish mutation (A53E) attenuates α-synuclein aggregation and membrane binding.
Biochemistry.
2014; 53(41): 6419–21. PubMed Abstract
| Publisher Full Text
- 158.
Rutherford NJ, Giasson BI:
The A53E α-synuclein pathological mutation demonstrates reduced aggregation propensity in vitro and in cell culture.
Neurosci Lett.
2015; 597: 43–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 159.
Porcari R, Proukakis C, Waudby CA, et al.:
The H50Q mutation induces a 10-fold decrease in the solubility of α-synuclein.
J Biol Chem.
2015; 290(4): 2395–404. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 160.
Fares MB, Ait-Bouziad N, Dikiy I, et al.:
The novel Parkinson's disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells.
Hum Mol Genet.
2014; 23(17): 4491–509. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 161.
Ferreon AC, Moran CR, Ferreon JC, et al.:
Alteration of the alpha-synuclein folding landscape by a mutation related to Parkinson's disease.
Angew Chem Int Ed Engl.
2010; 49(20): 3469–72. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 162.
Rospigliosi CC, McClendon S, Schmid AW, et al.:
E46K Parkinson's-linked mutation enhances C-terminal-to-N-terminal contacts in alpha-synuclein.
J Mol Biol.
2009; 388(5): 1022–32. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 163.
Bussell R Jr, Eliezer D:
Residual structure and dynamics in Parkinson's disease-associated mutants of alpha-synuclein.
J Biol Chem.
2001; 276(49): 45996–6003. PubMed Abstract
| Publisher Full Text
- 164.
Xu L, Ma B, Nussinov R, et al.:
Familial Mutations May Switch Conformational Preferences in α-Synuclein Fibrils.
ACS Chem Neurosci.
2017. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 165.
Sahay S, Ghosh D, Dwivedi S, et al.:
Familial Parkinson disease-associated mutations alter the site-specific microenvironment and dynamics of α-synuclein.
J Biol Chem.
2015; 290(12): 7804–22. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 166.
Acharya S, Saha S, Ahmad B, et al.:
Effects of Mutations on the Reconfiguration Rate of α-Synuclein.
J Phys Chem B.
2015; 119(50): 15443–50. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 167.
Sanjeev A, Kumar Mattaparthi V:
Computational Investigation on Tyrosine to Alanine Mutations Delaying the Early Stage of α-Synuclein Aggregation.
Curr Proteomics.
2017; 14(1): 31–41. Publisher Full Text
| Faculty Opinions Recommendation
- 168.
Chen SW, Drakulic S, Deas E, et al.:
Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation.
Proc Natl Acad Sci U S A.
2015; 112(16): E1994–2003. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 169.
Xin W, Emadi S, Williams S, et al.:
Toxic Oligomeric Alpha-Synuclein Variants Present in Human Parkinson's Disease Brains Are Differentially Generated in Mammalian Cell Models.
Biomolecules.
2015; 5(3): 1634–51. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 170.
Paslawski W, Mysling S, Thomsen K, et al.:
Co-existence of two different α-synuclein oligomers with different core structures determined by hydrogen/deuterium exchange mass spectrometry.
Angew Chem Int Ed Engl.
2014; 53(29): 7560–3. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 171.
Stefanovic AN, Lindhoud S, Semerdzhiev SA, et al.:
Oligomers of Parkinson's Disease-Related α-Synuclein Mutants Have Similar Structures but Distinctive Membrane Permeabilization Properties.
Biochemistry.
2015; 54(20): 3142–50. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 172.
Gustot A, Gallea JI, Sarroukh R, et al.:
Amyloid fibrils are the molecular trigger of inflammation in Parkinson's disease.
Biochem J.
2015; 471(3): 323–33. PubMed Abstract
| Publisher Full Text
- 173.
Deas E, Cremades N, Angelova PR, et al.:
Alpha-Synuclein Oligomers Interact with Metal Ions to Induce Oxidative Stress and Neuronal Death in Parkinson's Disease.
Antioxid Redox Signal.
2016; 24(7): 376–91. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 174.
Oikawa T, Nonaka T, Terada M, et al.:
α-Synuclein Fibrils Exhibit Gain of Toxic Function, Promoting Tau Aggregation and Inhibiting Microtubule Assembly.
J Biol Chem.
2016; 291(29): 15046–56. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 175.
Bhak G, Lee J, Kim T, et al.:
Molecular inscription of environmental information into protein suprastructures: temperature effects on unit assembly of α-synuclein oligomers into polymorphic amyloid fibrils.
Biochem J.
2014; 464(2): 259–69. PubMed Abstract
| Publisher Full Text
- 176.
Bousset L, Pieri L, Ruiz-Arlandis G, et al.:
Structural and functional characterization of two alpha-synuclein strains.
Nat Commun.
2013; 4: 2575. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 177.
Gath J, Bousset L, Habenstein B, et al.:
Unlike twins: an NMR comparison of two α-synuclein polymorphs featuring different toxicity.
PLoS One.
2014; 9(3): e90659. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 178.
Makky A, Bousset L, Polesel-Maris J, et al.:
Nanomechanical properties of distinct fibrillar polymorphs of the protein α-synuclein.
Sci Rep.
2016; 6: 37970. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 179.
Melki R:
Role of Different Alpha-Synuclein Strains in Synucleinopathies, Similarities with other Neurodegenerative Diseases.
J Parkinsons Dis.
2015; 5(2): 217–27. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 180.
Paravastu AK, Leapman RD, Yau WM, et al.:
Molecular structural basis for polymorphism in Alzheimer's beta-amyloid fibrils.
Proc Natl Acad Sci U S A.
2008; 105(47): 18349–54. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 181.
Petkova AT, Leapman RD, Guo Z, et al.:
Self-propagating, molecular-level polymorphism in Alzheimer's beta-amyloid fibrils.
Science.
2005; 307(5707): 262–5. PubMed Abstract
| Publisher Full Text
- 182.
Makarava N, Savtchenko R, Alexeeva I, et al.:
New Molecular Insight into Mechanism of Evolution of Mammalian Synthetic Prions.
Am J Pathol.
2016; 186(4): 1006–14. PubMed Abstract
| Publisher Full Text
- 183.
Sano K, Atarashi R, Ishibashi D, et al.:
Conformational properties of prion strains can be transmitted to recombinant prion protein fibrils in real-time quaking-induced conversion.
J Virol.
2014; 88(20): 11791–801. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 184.
Solforosi L, Milani M, Mancini N, et al.:
A closer look at prion strains: characterization and important implications.
Prion.
2013; 7(2): 99–108. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 185.
Morales R, Abid K, Soto C:
The prion strain phenomenon: molecular basis and unprecedented features.
Biochim Biophys Acta.
2007; 1772(6): 681–91. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 186.
Wickner RB:
Yeast and Fungal Prions.
Cold Spring Harb Perspect Biol.
2016; 8(9): pii: a023531. PubMed Abstract
| Publisher Full Text
- 187.
Sharma A, Bruce KL, Chen B, et al.:
Contributions of the Prion Protein Sequence, Strain, and Environment to the Species Barrier.
J Biol Chem.
2016; 291(3): 1277–88. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 188.
Toyama BH, Kelly MJ, Gross JD, et al.:
The structural basis of yeast prion strain variants.
Nature.
2007; 449(7159): 233–7. PubMed Abstract
| Publisher Full Text
- 189.
Arvidsson S, Pilebro B, Westermark P, et al.:
Amyloid Cardiomyopathy in Hereditary Transthyretin V30M Amyloidosis - Impact of Sex and Amyloid Fibril Composition.
PLoS One.
2015; 10(11): e0143456. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 190.
Sneideris T, Darguzis D, Botyriute A, et al.:
pH-Driven Polymorphism of Insulin Amyloid-Like Fibrils.
PLoS One.
2015; 10(8): e0136602. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 191.
Kodali R, Williams AD, Chemuru S, et al.:
Abeta(1–40) forms five distinct amyloid structures whose beta-sheet contents and fibril stabilities are correlated.
J Mol Biol.
2010; 401(3): 503–17. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 192.
Morales R, Callegari K, Soto C:
Prion-like features of misfolded Aβ and tau aggregates.
Virus Res.
2015; 207: 106–12. PubMed Abstract
| Publisher Full Text
- 193.
Peelaerts W, Bousset L, Van der Perren A, et al.:
α-Synuclein strains cause distinct synucleinopathies after local and systemic administration.
Nature.
2015; 522(7556): 340–4. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 194.
Lerner A, Bagic A:
Olfactory pathogenesis of idiopathic Parkinson disease revisited.
Mov Disord.
2008; 23(8): 1076–84. PubMed Abstract
| Publisher Full Text
- 195.
Visanji NP, Brooks PL, Hazrati LN, et al.:
The prion hypothesis in Parkinson's disease: Braak to the future.
Acta Neuropathol Commun.
2013; 1: 2. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 196.
Ulusoy A, Musgrove RE, Rusconi R, et al.:
Neuron-to-neuron α-synuclein propagation in vivo is independent of neuronal injury.
Acta Neuropathol Commun.
2015; 3: 13. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 197.
Tofaris GK, Goedert M, Spillantini MG:
The Transcellular Propagation and Intracellular Trafficking of α-Synuclein.
Cold Spring Harb Perspect Med.
2016; pii: a024380. PubMed Abstract
| Publisher Full Text
- 198.
Mao X, Ou MT, Karuppagounder SS, et al.:
Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3.
Science.
2016; 353(6307): pii: aah3374. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 199.
Illes-Toth E, Ramos MR, Cappai R, et al.:
Distinct higher-order α-synuclein oligomers induce intracellular aggregation.
Biochem J.
2015; 468(3): 485–93. PubMed Abstract
| Publisher Full Text
- 200.
Schlüter H, Apweiler R, Holzhütter H, et al.:
Finding one's way in proteomics: a protein species nomenclature.
Chem Cent J.
2009; 3: 11. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 201.
Smith LM, Kelleher NL:
Proteoform: a single term describing protein complexity.
Nat Methods.
2013; 10(3): 186–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 202.
Uversky VN:
p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure-Function Continuum Concept.
Int J Mol Sci.
2016; 17(11): pii: E1874. PubMed Abstract
| Publisher Full Text
| Free Full Text
Comments on this article Comments (0)