Bhowmick R and Hickson ID. The "enemies within": regions of the genome that are inherently difficult to replicate [version 1; peer review: 2 approved]. F1000Research 2017, 6(F1000 Faculty Rev):666 (https://doi.org/10.12688/f1000research.11024.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
1Center for Chromosome Stability, Department of Cellular and Molecular Medicine, Panum Institute, University of Copenhagen, Blegdamsvej 3B, 2200 Copenhagen N, Denmark
OPEN PEER REVIEW
REVIEWER STATUS
Abstract
An unusual feature of many eukaryotic genomes is the presence of regions that appear intrinsically difficult to copy during the process of DNA replication. Curiously, the location of these difficult-to-replicate regions is often conserved between species, implying a valuable role in some aspect of genome organization or maintenance. The most prominent class of these regions in mammalian cells is defined as chromosome fragile sites, which acquired their name because of a propensity to form visible gaps/breaks on otherwise-condensed chromosomes in mitosis. This fragility is particularly apparent following perturbation of DNA replication—a phenomenon often referred to as “replication stress”. Here, we review recent data on the molecular basis for chromosome fragility and the role of fragile sites in the etiology of cancer. In particular, we highlight how studies on fragile sites have provided unexpected insights into how the DNA repair machinery assists in the completion of DNA replication.
Keywords
DNA replication, genome, DNA repair machinery, chromosome fragility, replication stress
Corresponding author:
Ian D Hickson
Competing interests:
The authors declare that they have no competing interests.
Grant information:
Work in the authors’ laboratory is supported by the Danish National Research Foundation (DNRF115), the Nordea Foundation, and the European Research Council. RB acknowledges the support of a postdoctoral fellowship from the Danish Medical Research Council.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The duplication of the genome during the process of DNA replication is fundamental for the viability of all cells. This process initiates from DNA replication origins and proceeds along the parental DNA template in a bidirectional manner until convergence with another replication fork occurs (“DNA replication termination”)1. DNA replication generally occurs with remarkable accuracy. For example, it is estimated that only one error occurs for every 100 million base pairs of replicated DNA in human cells, a degree of fidelity that is achieved by a combination of accurate copying of the template coupled with efficient error correction mechanisms2. Nevertheless, any event that interferes with the DNA replication process has the potential to generate errors that can be inherited by future cell generations. The perturbation of DNA replication in this way is broadly termed “replication stress”3. Although there are several definitions of replication stress, we refer to it as a perturbation of replication fork progression leading to fork stalling and the need for fork protection/repair processes to be employed. An array of highly coordinated DNA repair mechanisms have evolved to ensure that the completion of DNA replication occurs in a timely and faithful manner in order to maintain genomic integrity in the face of replication stress4,5. Defects in these protective functions can lead to genomic instability and, in multicellular organisms, to cancer predisposition6.
DNA replication stress is implicated as a major driver of tumorigenesis. This is because one of the key features of tumorigenesis is the activation or overexpression of proto-oncogenes, which drives cell proliferation by interfering with the regulatory pathways of cell cycle progression7. Uncontrolled cell proliferation interferes with replication timing and replication fork progression, manifesting as enhanced DNA replication stress8. This can generate chromosome rearrangements and mis-segregation during cell division9, which in turn can lead to aneuploidy (an abnormal number of chromosomes), which is a common feature of solid tumors10. Constitutive activation of a so-called DNA damage response (DDR) during oncogene-induced replication stress was the first evidence to link DNA replication stress with cancer11,12. This association was strengthened by the detection of mutations in DDR pathway genes in primary cancers13–15. Moreover, perturbation of DNA replication using agents that slow replication fork progression by inhibiting DNA polymerases (for example, aphidicolin [APH]) can generate DNA rearrangements, such as micro-deletions, that closely resemble those found in human tumors16. Strikingly, recent cancer genome-sequencing projects have revealed that more than half of the DNA rearrangements in cancers cluster within certain chromosomal loci known as common fragile sites (CFSs)17, including in the FHIT gene located within FRA3B and the WWOX gene within FRA16D. In this article, we discuss recent advances in our knowledge of replication stress and how it particularly affects CFSs.
Features of common fragile sites
CFSs are regions of the human genome that are present in all individuals18. They were first described over three decades ago as sites where gaps or constrictions are visible in condensed mitotic chromosomes19. Formation of these gaps/constrictions (usually termed fragile site “expression”) is far more prominent in cells exposed to mild replication stress. CFSs are conserved to a large extent in mice20–23 and other primates24. Over 200 CFSs have been identified in human lymphocytes; however, it is known that the frequency of expression of any single CFS depends on the nature of the replication stress and the cell type. As a result, most of the known sites are infrequently expressed25. Sequence analysis of CFSs has revealed some conserved features. For example, most commonly expressed CFSs harbor a very long gene that takes more than one cell cycle to transcribe. This led to the hypothesis that an underlying cause of CFS expression is the inevitable collision between the replication fork and the transcription machinery that must occur in each S-phase26. Another feature shared in some cases is the presence of stretches of interrupted AT-dinucleotide repeats that influence DNA helix flexibility27,28 and have the potential to form stable DNA secondary structures29. It was recently demonstrated that replication forks pause at CFS loci in the absence of proteins involved in their maintenance, likely due to the accumulation of DNA-associated structures such as R-loops30. This is proposed to lead to replication fork arrest and DNA breakage. In cultured cells, CFS expression is generally triggered by the use of low doses of APH to induce mild replication stress31,32. This treatment also induces sister chromatid exchange and an elevated frequency of translocations and deletions at CFSs33. In the context of cancer, CFSs are hotspots for micro-deletions and are associated with chromosomal translocation breakpoints34–38. CFSs also act as hotspots for viral DNA integration, which can lead to cancer development39–42. Despite these observations, it is clear that many of the structural changes observed at CFSs in cancers are present on only one allele, and hence these have been proposed to be passenger, not driver, mutations33,37,38. Nevertheless, some mouse models with CFS gene inactivation have shown an increased tumor burden34,35.
Common fragile site maintenance pathways
Many proteins involved in the recognition and/or repair of DNA damage triggered by replication stress have been reported to play a role in CFS maintenance. These include the ATR and CHK1 kinases, BRCA1, RAD51, and FANCD230,43–45. A major challenge for CFS maintenance systems in cells undergoing replication stress is ensuring that the completion of DNA replication occurs before the cell enters mitosis. CFSs appear to replicate in late S-phase, which may render them more susceptible to DNA replication stress46. Indeed, they can remain under-replicated and escape the checkpoint surveillance, even when the cell enters mitosis47. This is potentially dangerous, as it can enhance the formation of chromatin bridges, ultra-fine anaphase DNA bridges (UFBs), and lagging chromosomes during mitosis. Anaphase bridges, in turn, can promote the nondisjunction of sister chromatids and micronucleus formation47–49. Not surprisingly, therefore, cells have evolved efficient mechanisms to process anaphase bridges and counteract mitotic defects. The mechanism of this resolution is poorly characterized but involves the BLM helicase in association with topoisomerase IIIα, RMI1 and RMI2, and the Plk1-interacting checkpoint helicase (PICH)49,50.
Common fragile site replication is completed in mitosis during stress conditions
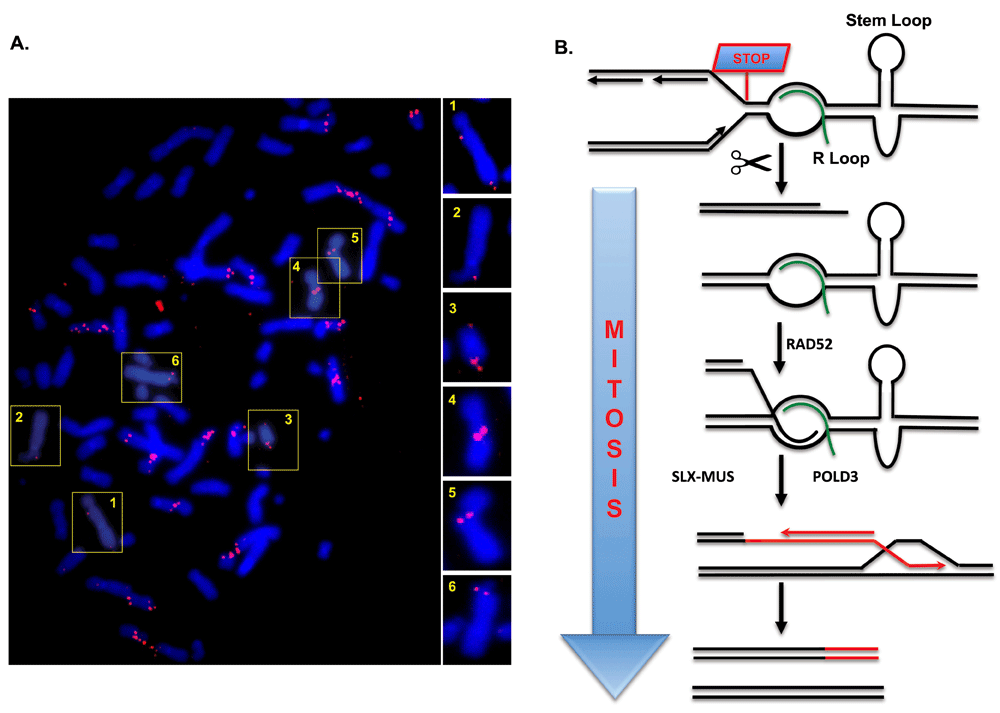
Though seemingly counterintuitive, CFS expression is a protective mechanism to promote CFS maintenance44. This can be explained by the fact that CFS expression is not accidental but instead is a programmed event mediated by DNA repair proteins such as the MUS81-EME1 endonuclease48,51. This finding led us to investigate whether cells might be attempting to rescue failed replication at CFSs in mitosis. Indeed, we demonstrated that replication stress activates nascent DNA synthesis at CFSs in the prophase of mitosis (Figure 1)47. Interestingly, this synthesis coincides with the sites of the gaps/breaks in the condensed chromosomes. Thus, it would appear that CFS expression denotes sites where DNA synthesis is still ongoing in mitosis. This mitotic DNA synthesis, which we termed MiDAS, has also been detected in other studies using a different kind of replication stress52 and in a different cell type53. Thus, MiDAS is likely to be a universal mechanism used by cells in mitosis to buffer the effects of DNA replication stress.
Figure 1. Mitotic DNA synthesis (MiDAS) occurs via a break-induced replication (BIR)-like process.
(A) Metaphase spread of U2OS cells treated with low-dose aphidicolin showing mitotic 5-ethynyl-2′-deoxyuridine (EdU) incorporation. Most sites of EdU incorporation exhibit a conservative pattern of DNA synthesis, having EdU incorporation on only one sister chromatid. Selected chromosomes are shown in numbered boxes and are enlarged on the right. (B) Model showing how MiDAS might occur via a BIR-like process. A replication fork stalls at a common fragile site (CFS), perhaps due to the presence of an R-loop or a DNA secondary structure (a stem-loop structure is shown as an example). The fork is then cleaved by an endonuclease, followed by limited end resection of the generated DNA end. This exposes a region of micro-homology that can be annealed with the partially single-stranded template DNA by the RAD52 protein. Processing of the resulting replication intermediate by the activated SLX-MUS complex (SLX4 in complex with MUS81-EME1 and other nucleases) in early mitosis is then associated with POLD3-dependent conservative DNA repair synthesis. This process would account for the high level of copy number variations that arise at CFS loci in cancer cells. For clarity, the replication fork merging with the MiDAS bubble from the right is omitted. If the converging fork were to suffer the same fate as the fork depicted, this could lead to the newly synthesized DNA occurring on both sister chromatids.
On the basis of our observations, we propose that MiDAS is not just a continuation of normal semi-conservative DNA replication but rather is a form of DNA repair analogous to break-induced replication (BIR) that has been characterized largely in budding yeast. BIR is generally a conservative form of DNA synthesis, and the nascent DNA is present on only one sister chromatid, leading to the accumulation of changes/mutations in only one allele, as is commonly found at CFSs in cancers33,37,38,54 (Figure 1). Consistent with this, we have observed characteristic patterns of DNA replication that resemble this in mitotic human cells. MiDAS also requires the RAD52 and POLD3 proteins, the homologs of which (Rad52 and Pol32, respectively) are essential for BIR in yeast47,54–56. Indeed, a recent study showed that RAD52 is required more generally for repairing perturbed DNA replication forks in cells exposed to replication stress57. Interestingly, MiDAS in human cells does not require RAD5154, although most BIR in yeast is Rad51-dependent56.
Closing remarks
It is curious that MiDAS in human cells is apparently RAD51-independent but that canonical BIR in yeast requires the DNA strand invasion function of this protein. This suggests that MiDAS might represent an atypical form of BIR that occurs in mitosis only at a time when BRCA2 and RAD51 are excluded from the chromatin. One possible explanation for this comes from studies in yeast indicating that RAD51-independent BIR in that organism requires much less homology for DNA copying to be initiated58. As MiDAS occurs during a narrow time window in early mitosis and involves sister chromatids that are already in very close proximity, there might be a selective advantage in using a “quick and dirty” form of repair that serves to prevent the accumulation of fatal mitotic abnormalities at the expense of mutations.
There is still much to be understood about MiDAS; for example, how is the reaction initiated if the replisome remains associated with stalled forks in the CFS loci, and how similar is the mechanism of BIR in human cells to that defined in yeast? Intriguingly, BIR has been proposed to be required for the maintenance of telomeres in cells lacking telomerase, and therefore MiDAS might be mechanistically related to telomere maintenance by the so-called ALT (alternative lengthening of telomeres) pathway59. Moreover, it is possible that the existence of MiDAS could be exploited as a therapeutic approach to kill cancer cells, as MiDAS inhibition is synergistically toxic to cancer cells in combination with inhibitors of ATR kinase54.
Research on CFSs has been ongoing for decades, but these conserved “enemies within the genome” still manage to provide new surprises and insights into the biology of chromosome maintenance. It will be intriguing to define why CFSs have not been eliminated during evolution. Their conservation predicts a positive role in DNA metabolism, but this role remains elusive. Clearly, there is still much to be learned from studying these idiosyncratic regions of our genome.
Competing interests
The authors declare that they have no competing interests.
Grant information
Work in the authors’ laboratory is supported by the Danish National Research Foundation (DNRF115), the Nordea Foundation, and the European Research Council. RB acknowledges the support of a postdoctoral fellowship from the Danish Medical Research Council.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We thank Hocine Mankouri for helpful comments on the manuscript.
Faculty Opinions recommended
References
1.
Fragkos M, Ganier O, Coulombe P, et al.:
DNA replication origin activation in space and time.
Nat Rev Mol Cell Biol.
2015; 16(6): 360–74. PubMed Abstract
| Publisher Full Text
7.
Hills SA, Diffley JF:
DNA replication and oncogene-induced replicative stress.
Curr Biol.
2014; 24(10): R435–44. PubMed Abstract
| Publisher Full Text
8.
Mazouzi A, Velimezi G, Loizou JI:
DNA replication stress: causes, resolution and disease.
Exp Cell Res.
2014; 329(1): 85–93. PubMed Abstract
| Publisher Full Text
13.
Curtin NJ:
DNA repair dysregulation from cancer driver to therapeutic target.
Nat Rev Cancer.
2012; 12(12): 801–17. PubMed Abstract
| Publisher Full Text
16.
Durkin SG, Ragland RL, Arlt MF, et al.:
Replication stress induces tumor-like microdeletions in FHIT/FRA3B.
Proc Natl Acad Sci U S A.
2008; 105(1): 246–51. PubMed Abstract
| Publisher Full Text
| Free Full Text
17.
Burrow AA, Williams LE, Pierce LC, et al.:
Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites.
BMC Genomics.
2009; 10: 59. PubMed Abstract
| Publisher Full Text
| Free Full Text
20.
Glover TW, Hoge AW, Miller DE, et al.:
The murine Fhit gene is highly similar to its human orthologue and maps to a common fragile site region.
Cancer Res.
1998; 58(15): 3409–14. PubMed Abstract
21.
Shiraishi T, Druck T, Mimori K, et al.:
Sequence conservation at human and mouse orthologous common fragile regions, FRA3B/FHIT and Fra14A2/Fhit.
Proc Natl Acad Sci U S A.
2001; 98(10): 5722–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
22.
Krummel KA, Denison SR, Calhoun E, et al.:
The common fragile site FRA16D and its associated gene WWOX are highly conserved in the mouse at Fra8E1.
Genes Chromosomes Cancer.
2002; 34(2): 154–67. PubMed Abstract
| Publisher Full Text
23.
Rozier L, El-Achkar E, Apiou F, et al.:
Characterization of a conserved aphidicolin-sensitive common fragile site at human 4q22 and mouse 6C1: possible association with an inherited disease and cancer.
Oncogene.
2004; 23(41): 6872–80. PubMed Abstract
| Publisher Full Text
24.
Ruiz-Herrera A, Garcia F, Frönicke L, et al.:
Conservation of aphidicolin-induced fragile sites in Papionini (Primates) species and humans.
Chromosome Res.
2004; 12(7): 683–90. PubMed Abstract
| Publisher Full Text
25.
Whellan DJ, Ellis SJ, Kraus WE, et al.:
Method for establishing authorship in a multicenter clinical trial.
Ann Intern Med.
2009; 151(6): 414–20. PubMed Abstract
| Publisher Full Text
| Free Full Text
28.
Mishmar D, Rahat A, Scherer SW, et al.:
Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site.
Proc Natl Acad Sci U S A.
1998; 95(14): 8141–6. PubMed Abstract
| Free Full Text
31.
Ikegami S, Taguchi T, Ohashi M, et al.:
Aphidicolin prevents mitotic cell division by interfering with the activity of DNA polymerase-alpha.
Nature.
1978; 275(5679): 458–60. PubMed Abstract
32.
Cheng CH, Kuchta RD:
DNA polymerase epsilon: aphidicolin inhibition and the relationship between polymerase and exonuclease activity.
Biochemistry.
1993; 32(33): 8568–74. PubMed Abstract
| Publisher Full Text
33.
Arlt MF, Durkin SG, Ragland RL, et al.:
Common fragile sites as targets for chromosome rearrangements.
DNA Repair (Amst).
2006; 5(9–10): 1126–35. PubMed Abstract
| Publisher Full Text
34.
Hellman A, Zlotorynski E, Scherer SW, et al.:
A role for common fragile site induction in amplification of human oncogenes.
Cancer Cell.
2002; 1(1): 89–97. PubMed Abstract
| Publisher Full Text
35.
Kotzot D, Martinez MJ, Bagci G, et al.:
Parental origin and mechanisms of formation of cytogenetically recognisable de novo direct and inverted duplications.
J Med Genet.
2000; 37(4): 281–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
36.
Miller CT, Lin L, Casper AM, et al.:
Genomic amplification of MET with boundaries within fragile site FRA7G and upregulation of MET pathways in esophageal adenocarcinoma.
Oncogene.
2006; 25(3): 409–18. PubMed Abstract
| Publisher Full Text
37.
Hecht F, Hecht BK:
Fragile sites and chromosome breakpoints in constitutional rearrangements I. Amniocentesis.
Clin Genet.
1984; 26(3): 169–73. PubMed Abstract
| Publisher Full Text
38.
Yunis JJ:
Fragile sites and predisposition to leukemia and lymphoma.
Cancer Genet Cytogenet.
1984; 12(1): 85–8. PubMed Abstract
| Publisher Full Text
39.
Matovina M, Sabol I, Grubisić G, et al.:
Identification of human papillomavirus type 16 integration sites in high-grade precancerous cervical lesions.
Gynecol Oncol.
2009; 113(1): 120–7. PubMed Abstract
| Publisher Full Text
40.
Thorland EC, Myers SL, Gostout BS, et al.:
Common fragile sites are preferential targets for HPV16 integrations in cervical tumors.
Oncogene.
2003; 22(8): 1225–37. PubMed Abstract
| Publisher Full Text
41.
Thorland EC, Myers SL, Persing DH, et al.:
Human papillomavirus type 16 integrations in cervical tumors frequently occur in common fragile sites.
Cancer Res.
2000; 60(21): 5916–21. PubMed Abstract
42.
Yu T, Ferber MJ, Cheung TH, et al.:
The role of viral integration in the development of cervical cancer.
Cancer Genet Cytogenet.
2005; 158(1): 27–34. PubMed Abstract
| Publisher Full Text
45.
Turner BC, Ottey M, Zimonjic DB, et al.:
The fragile histidine triad/common chromosome fragile site 3B locus and repair-deficient cancers.
Cancer Res.
2002; 62(14): 4054–60. PubMed Abstract
46.
Le Beau MM, Rassool FV, Neilly ME, et al.:
Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: implications for the mechanism of fragile site induction.
Hum Mol Genet.
1998; 7(4): 755–61. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
50.
Naim V, Rosselli F:
The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities.
Nat Cell Biol.
2009; 11(6): 761–8. PubMed Abstract
| Publisher Full Text
51.
Naim V, Wilhelm T, Debatisse M, et al.:
ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis.
Nat Cell Biol.
2013; 15(8): 1008–15. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
58.
Ira G, Haber JE:
Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences.
Mol Cell Biol.
2002; 22(18): 6384–92. PubMed Abstract
| Publisher Full Text
| Free Full Text
1
Center for Chromosome Stability, Department of Cellular and Molecular Medicine, Panum Institute, University of Copenhagen, Blegdamsvej 3B, 2200 Copenhagen N, Denmark
Work in the authors’ laboratory is supported by the Danish National Research Foundation (DNRF115), the Nordea Foundation, and the European Research Council. RB acknowledges the support of a postdoctoral fellowship from the Danish Medical Research Council.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Bhowmick R and Hickson ID. The "enemies within": regions of the genome that are inherently difficult to replicate [version 1; peer review: 2 approved]. F1000Research 2017, 6(F1000 Faculty Rev):666 (https://doi.org/10.12688/f1000research.11024.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
Noguchi E. Reviewer Report For: The "enemies within": regions of the genome that are inherently difficult to replicate [version 1; peer review: 2 approved]. F1000Research 2017, 6(F1000 Faculty Rev):666 (https://doi.org/10.5256/f1000research.11891.r22177)
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Noguchi E. Reviewer Report For: The "enemies within": regions of the genome that are inherently difficult to replicate [version 1; peer review: 2 approved]. F1000Research 2017, 6(F1000 Faculty Rev):666 (https://doi.org/10.5256/f1000research.11891.r22177)
Schildkraut C and Madireddy A. Reviewer Report For: The "enemies within": regions of the genome that are inherently difficult to replicate [version 1; peer review: 2 approved]. F1000Research 2017, 6(F1000 Faculty Rev):666 (https://doi.org/10.5256/f1000research.11891.r22176)
We confirm that we have read this submission and believe that we have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
We confirm that we have read this submission and believe that we have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Schildkraut C and Madireddy A. Reviewer Report For: The "enemies within": regions of the genome that are inherently difficult to replicate [version 1; peer review: 2 approved]. F1000Research 2017, 6(F1000 Faculty Rev):666 (https://doi.org/10.5256/f1000research.11891.r22176)
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)