Introduction
The interleukin-17 (IL-17) cytokine family is composed of six defined members, including IL-17A through IL-17F1. Among the IL-17 family members, IL-17A and IL-17F have the best-characterized proinflammatory activity. Although the genes encoding IL-17A and IL-17F are both located on chromosome 1 and 6 (respectively), in mice and humans2, their functions can be similar or distinct, depending on the type of infection3. Although other members of the IL-17 family such as IL-17B, IL-17C, and IL-17D can also induce the production of proinflammatory cytokines and chemokines4, their functions are not as well characterized and will be only briefly summarized. The IL-17 cytokine family employs various cytokine receptors (IL-17RA, IL-17RB, IL-17RC, IL-17RD, and IL-17RE) on target cells to mediate their biological functions5. IL-17R is a heteromeric receptor comprising IL-17RA and IL-17RC and mediates signaling of IL-17A and IL-17F. In contrast, partnering of IL-17RA with IL-17RB is thought to mediate IL-17E signaling whereas IL-17RA partnering with IL-17RE mediates IL-17C signaling5. IL-17Rs are ubiquitously expressed in various cell types ranging from leukocytes to fibroblasts, epithelial cells, mesothelial cells, endothelial cells, and keratinocytes6,7. IL-17A or IL-17F mediates their biological function through the IL-17R via the activation of nuclear factor-kappa B (NF-κB) and mitogen-activated protein kinase (MAPK), leading to the production of proinflammatory cytokines and chemokines8–11. Tumor necrosis factor receptor-associated factor 6 (TRAF-6) plays an indispensable role in IL-17R signaling as IL-17 stimulation fails to activate IL-17R signaling in TRAF-6-deficient mouse embryonic fibroblasts12,13. In addition, NF-κB activator 1 (Act1) is important for IL-17R signaling, where it acts as an adapter molecule for the recruitment of TRAF-6 with IL-17R14,15. In this review, we will use IL-17 to refer to IL-17A.
Upon exposure to pathogen or pathogen-associated molecular patterns (PAMPs), dendritic cells, monocytes, and macrophages induce cytokines such as IL-23, IL-1β, IL-6, and transforming growth factor-beta (TGF-β), which initiate the differentiation and polarization of naïve CD4+ T cells toward the T helper cell type 17 (Th17) subsets16. Low levels of TGF-β support induction of the transcription factor RAR-related orphan receptor gamma (RORγ) and differentiation toward a Th17 subset, while high levels of TGF-β along with defined cytokines such as IL-2 mediate transition to regulatory T cells (Tregs) through the activation of the transcription factor, fork head box P3 (Foxp3)17–19. Th17 cells are considered a primary source of IL-17 and co-produce other cytokines, including IL-22, IL-21, tumor necrosis factor-alpha (TNF-α), and granulocyte macrophage-colony-stimulating factor (GM-CSF)20,21. However, depending on the cytokine milieu, Th17 cells can exhibit substantial plasticity in cytokine production22. Th17 cells can also co-express GATA binding protein 3 (GATA-3) or T-box transcription factor (T-bet), allowing them to progress into either IL-4-expressing or interferon-gamma (IFN-γ)-expressing Th17 subsets23. Thus, it is likely that during infection in vivo, Th17 cells exhibit substantial plasticity and can co-express Th17 cytokines along with other Th1, Th2, and Treg-associated cytokines. Additionally, in response to early IL-23 and IL-1β production by myeloid cells, innate cells such as γδ T cells24 and group 3 innate lymphoid cells (iLC3)25,26 can produce IL-17 and mediate early immune responses. Other immune cells such as neutrophils27–29, invariant natural killer T (iNKT)30 cells, innate Th17 cells (iTh17)31, and natural killer (NK)32 cells can also produce IL-17 through stimulation of TGF-β, IL-1β, IL-6, IL-23, or alpha-galactoceramide (α-galcer)33. A primary mechanism by which IL-17 mediates protection against pathogens (such as Klebsiella, Candida, and Chlamydia) is through the induction of chemokines and cytokines and downstream recruitment of neutrophils34,35. IL-17 can act alone or in synergy with other cytokines such as TNF-α and IL-22 to mediate induction of neutrophil-recruiting chemokines such as granulocyte-colony-stimulating factor (G-CSF) and C-X-C motif chemokine ligand 1 (CXCL1) and regulate neutrophil-mediated destruction of pathogens36. In addition, IL-17 alone or synergistically with IL-22 or 1,25-dihydroxyvitamin D3 induces the expression of anti-microbial proteins such as Lipocalin-237, β-defensin38, S100A7 (psoriasin), S100A8/9 (calprotectin), and cathelicidin (LL37), resulting in pathogen control11, likely through direct anti-microbial actions. Our recent knowledge on the role of IL-17 in immunity to various pathogens, including extracellular39,40 or intracellular41–43 bacteria, fungi9,44, viruses45, and parasites46,47, has emerged within the past decade. In this short review, we will summarize the recent progress in the field of IL-17-mediated immune responses against various infections.
Role of IL-17 in immunity to extracellular bacterial infection
The role of IL-17 in host defense against extracellular bacteria is thought to be primarily through the induction of anti-microbial molecules and mediation of neutrophil recruitment at the site of infection guided by chemokine gradients. Early studies with IL-17R-deficient mice demonstrated a critical role for IL-17 in the clearance of the extracellular pulmonary pathogen Klebsiella pneumoniae infection. IL-17R-deficient mice upon infection with K. pneumoniae produced lower levels of the neutrophil-driving cytokine G-CSF and neutrophil-recruiting chemokine, macrophage inflammatory protein-2 (MIP-2). These changes in cytokines and chemokines in IL-17R-deficient mice resulted in decreased neutrophil infiltration into the lung and subsequently higher bacterial burden along with increased mortality48. Additionally, IL-17R-deficient mice are more susceptible to a variety of mucosal extracellular pathogens, including the gut-specific pathogen Citrobacter rodentium49, skin pathogen Staphylococcus aureus50, and pulmonary pathogen Bordetella pertussis51. Moreover, neutralization of IL-17 resulted in the suppression of anti-microbial peptide β-defensin production, which killed invading S. aureus at mucosal surfaces52. These studies provide the consensus that upon infection with extracellular pathogens, γδ T cells53, iLC354, and iNKT55 cells are important early producers of IL-17 which are associated with innate immunity following extracellular bacterial infections. In addition, Th17 cells are involved in the IL-17-mediated responses associated with adaptive immune responses56,57. Therefore, these studies suggest that induction of IL-17 and synchronized production of anti-microbial molecules and neutrophil recruitment help the resolution of extracellular infection. During extracellular pathogenesis, the major IL-17 responsive cell population is thought to be mucosal epithelial cells58,59. However, other studies suggest that macrophage or dendritic cells (or both) also express IL-17R and respond to IL-17 and downstream protective responses4,54. Recently, it was reported that innate immune defense against a highly antibiotic-resistant strain of K. pneumoniae depends on crosstalk between inflammatory monocytes and innate lymphocytes which is mediated by TNF-α and IL-1754. IL-17-producing resident epidermal γδ T cells are essential for protecting the host against a subsequent staphylococcal infection60. IL-17-dependent neutrophil-mediated protection is also observed during spontaneous S. aureus infection61,62 and K. pneumoniae infection63–65. Although in most studies IL-17 plays a protective role during extracellular bacterial infections, in some cases IL-17 can also mediate pathology associated with the infection. For example, the periodontal extracellular bacteria Porphyromonas gingivalis can directly promote autoimmune arthritis by the induction of Toll-like receptor 2 (TLR2)/IL-1Rα-driven IL-17 response in DBA/1J mice66. Furthermore, increased frequency of IL-17+ cells was observed in gingival tissue of patients with periodontitis67, likely produced by human CD4+ T cells68. Similarly, B. pertussis infection can bias the host immune response toward IL-17 production, which may be associated with cough pathology in pertussis infection56,69. Additionally, IL-17 is associated with the neutrophilia and airway inflammation during Haemophilus influenza infection in mice undergoing allergic airway disease70. Thus, IL-17 has an important role in protective immunity to extracellular pathogens through release of anti-microbial proteins from cell types such as epithelial cells and neutrophils (and monocytes). On the other hand, IL-17 induced in response to infection may mediate excessive inflammation and pathology.
Role of IL-17 in intracellular bacterial infection
Although infection by intracellular bacteria is predominantly cleared by Th1 immune responses, recent studies have described an emerging role for IL-17 in protection against intracellular pathogens such as Listeria monocytogenes71, Mycoplasma pneumonia72, Legionella pneumophila73,74, Salmonella typhimurium75, Chlamydia muridarum76, Francisella tularensis77, and Mycobacterium tuberculosis78. Following infection with intracellular pathogens, like infection with extracellular pathogens, both innate cells such as iLC-364 and γδ T cells79 and adaptive cells such as Th17 cells80 are the primary producers of IL-17. But during intracellular infection, unlike extracellular infection, macrophages or myeloid cells have been shown to be major responder cells to IL-17. In response to IL-17 stimulation, macrophages and myeloid cells secrete higher amounts of anti-microbial cytokines such as TNF-α, IFN-γ, or IL-12 and contribute to host immune response against infections such as F. tularensis77. Although γδ T cell-derived IL-17 has played a more prominent role in L. monocytogenes41, M. tuberculosis81, F. tularensis82, and Mycobacterium bovis Bacillus Calmette-Guérin83 infections, Th17 cells as well as CD8+ cells are also involved in the antigen-specific production of IL-17 at the site of infection84. In addition, IL-17-deficient mice experience higher bacterial burden associated with disorganized granuloma formation (reduced monocyte, granulocyte, and T cell recruitment within the granuloma) during infections with intracellular pathogens such as F. tularensis77, S. typhimurium85, or M. tuberculosis86. In some infection models, including C. muridarum, IL-17 complemented the protective role imparted by the IL-12/IFN-γ axis through the involvement of myeloid differentiation factor 88 (MyD88) signaling where MyD88-deficient infected mice showed reduced IL-17 responses along with reduced neutrophil infiltration, which is important for early control of disease pathogenesis87,88. However, excess IL-17 production is detrimental for the host, as IL-10-deficient mice exhibit increased mortality after pulmonary F. tularensis infection due to excessive inflammation induced by IL-1789, which suggests that IL-17 is tightly regulated by IL-10. However, other evidence suggests that the contribution of IL-17 may serve a more compensatory function under unfavorable conditions such as in the absence of type I and II interferon signaling, where a low-magnitude IL-17 response to L. monocytogenes or M. tuberculosis infection is evident87,90. On the contrary, early studies suggest that IL-17-mediated immunity is dispensable against M. tuberculosis infection as evident by the results obtained from either anti-IL-17 treated or IL-17R-deficient mice which were not more susceptible against infection with less virulent lab-adapted M. tuberculosis strains as compared with wild-type mice91,92. However, the involvement of IL-17 in mucosal vaccine-driven protection in murine models of tuberculosis seems to be crucial, as suggested by Gopal et al.93. IL-17-mediated induction of CXCL-9-11 is responsible for the recruitment of protective antigen-specific T cells as well as induction of CXCL-13 to localize C-X-C motif chemokine receptor 5 (CXCR5)-positive cytokine-producing T cells within lung granulomas of M. tuberculosis-infected mice94. Interestingly, IL-17 responses were involved in protection against a hyper-virulent clinical isolate M. tuberculosis HN878 strain, as IL-17-deficient mice infected with M. tuberculosis HN878 had significantly higher bacterial burden along with reduced chemokine expression and less organized granuloma formation95. However, there are some contradictory views regarding the role of IL-17 in the context of human tuberculosis. Some studies support the protective role of IL-17 during human tuberculosis as IL-17 helps in the generation of proinflammatory cytokines such as IL-12 and IFN-γ and restricts pathogenesis within the host96. In contrast, other reports identified that IL-17 had a negative correlation with tuberculosis treatment and disease outcome97. In addition, IL-17-producing T cells are reported to play an immunopathological role in patients with multidrug-resistant M. tuberculosis by promoting severe tissue damage, which may be associated with low effectiveness of the second-line drugs employed during treatment97. Moreover, IL-23-dependent IL-17 production is associated with neutrophil accumulation and inflammation during a chronic re-stimulation model of tuberculosis98. Indeed, exacerbated production of IL-17 appears to drive pathology by inducing S100A8/A9 proteins that recruit neutrophils into the lung99 and cause excessive inflammation in mice during tuberculosis. Therefore, at least in the context of tuberculosis, the M. tuberculosis strain to some extent specifically dictates the protective role of IL-17. Therefore, during intracellular pathogen infections, although IL-17 is mostly associated with host protection through regulation of chemokine and cytokine balance and infiltration of different immune cells to the site of infection, IL-17 activity should be tightly regulated in order to maintain the fine balance between protection and pathology induced by IL-17.
Role of IL-17 during sepsis
Although sepsis is a syndrome rather than a disease itself, the role for IL-17 in experimental murine sepsis models and human sepsis has been studied. In a colitis model, both IL-17-deficient mice and mice treated with IL-17 neutralizing antibody resulted in significant improvement in survival which was associated with reduced disease pathology and decreased bacteremia100,101. In line with this observation, IL-17 also drives sepsis-associated acute kidney injury by increasing the levels of proinflammatory cytokines and inducing neutrophil accumulation and tubular epithelial cell apoptosis102 in mouse models. More recently, targeting IL-17 has been shown to attenuate IL-18-dependent disease severity in a neonatal sepsis mouse model103. In vitro studies with the peripheral blood mononuclear cells (PBMCs) from healthy donors and patients undergoing severe sepsis showed increased Th17 cells in patients with sepsis when compared with healthy donors. Additionally, IL-17 neutralization increased IL-10 production in PBMCs, suggesting a role for IL-10 in modulating immune responses during sepsis104. Thus, IL-17 has a pathological role in sepsis, and targeting IL-17 may serve to resolve sepsis and sepsis-induced pathogenesis.
Role of IL-17 in parasitic infection
Although IL-17 has been considered an important player in the mediation of host protection against extracellular and some intracellular pathogens, the role of IL-17 in host defense against intracellular protozoan parasites remains less well studied. Infection studies demonstrate that Th17 cells mediate host defense against Trypanosoma cruzi105, Toxoplasma gondii106, Leishmania braziliensis107, and Echinococcus granulosus108 infections. NK cells are a major source of IL-17 during toxoplasmosis32. In addition, CD4+ and CD8+ cells express IL-17 in human toxoplasmosis and impact human pregnancy by controlling parasite invasion and replication which often cause fetal malfunction or abortion109. Increased IL-17 levels were detected in the PBMCs and tissue from leishmaniasis-infected patients and associated with enhanced neutrophil and macrophage-mediated destruction of the parasite110. Furthermore, IL-17R-deficient mice were associated with reduced production of the chemokine MIP-2 along with the suboptimal levels of neutrophil recruitment and higher parasitic load as compared with wild-type counterparts111. Additionally, during echinococcosis, IL-17 plays a crucial immune protective role by regulating the Tregs which are associated with tolerance during infection112. In contrast, in human cutaneous leishmaniasis113–115 and Eimeria tenella infection in chickens116, IL-17 contributed to the pathology through excessive inflammation and subsequent tissue damage. A recent report suggests that Leishmania guyanensis is associated with a cytoplasmic virus which enhances parasite virulence and is linked to increased IL-17 levels induced following L. guyanensis infection115. Neutralization of IL-17 was effective in reducing disease severity in a mouse model of cutaneous leishmaniasis, suggesting that IL-17 may have a strain-specific immunological role during leishmaniasis infection115. Despite having a protective role against T. gondii infection, IL-17 had a deleterious effect that is evident where neutralization of IL-17 had a partial protective role against the fatal disease117, through co-production of IL-10 and IFN-γ which regulated the exacerbated inflammation induced by IL-17. Taken together, these reports argue with previous reports and present new evidence in favor of the pathological role of IL-17 during parasitic infections. Therefore, during parasitic infection, the role of IL-17, whether protective or pathologic, has yet to be firmly established.
Role of IL-17 in fungal infection
IL-17 plays an immunologically important host protective role against fungal pathogens such as Candida albicans118, Cryptococcus neoformans119, Pneumocystis carinii120, and Aspergillus fumigatus121 in both humans and mice. Similar to the mechanisms seen in the intracellular and extracellular bacterial infections, fungal pathogens elicit IL-17 protective effects through the release of proinflammatory cytokines, chemokines, and anti-microbial peptides. During infection, IL-17 is expressed by various cell types, including oral resident γδ T cells122, iLC3123, and natural Th17 cells122. Moreover, the IL-17 cytokine family contributes in the development of NK cells which promote anti-fungal immunity by secreting GM-CSF, necessary for the fungicidal activity of neutrophils124,125. Recent advances in the field of oral candidiasis depict oral epithelial cells (OECs) as the major responder cells to IL-17 signaling126. These OECs produce β-defensin 3 through IL-17R signaling which is necessary for protection against oral candidiasis through both a neutrophil-dependent and -independent manner118. Caspase recruitment domain family member 9 (CARD-9) signaling is associated with the production of IL-17 during fungal infections127. Accordingly, humans with CARD-9128 or IL-17R deficiency have increased mucocandidiasis129 and are more vulnerable during systemic candidiasis124, and decreased IL-17 production is associated with increased susceptibility to fungal pathogens130. These studies suggest that fungal pathogens are dependent on IL-17-mediated recruitment of inflammatory cells for fungal control. In contrast, IL-17C subset is associated with lethal inflammation during candidiasis through induction of proinflammatory cytokines in renal epithelial cells131. Moreover, the IL-23/IL-17 pathway promotes inflammation and susceptibility to fungal infectious disease models such as C. albicans and A. fumigatus through excessive inflammation, which impairs anti-fungal resistance against those infections132–134. Therefore, critical observation on the particular role played by the IL-17 cytokine family is necessary before considering IL-17 signaling as a potential drug target.
Role of IL-17 in viral infection
Recent studies have addressed whether IL-17 is protective or pathologic in response to viral infections such as influenza (H1N1, H5N1), vaccinia virus, Epstein-Barr virus (EBV), herpes simplex virus (HSV), respiratory syncytial virus (RSV), human immunodeficiency virus (HIV), and hepatitis (B and C). Although several studies have suggested a protective role imparted by IL-17 signaling in host immunity during influenza infection, other studies have suggested a more pathological role instead. For example, it has been observed that depletion of IL-17 resulted in a more severe disease outcome in a mouse model of influenza, which was associated with increased weight loss as well as reduced survival135,136. Furthermore, adoptive transfer of Th17 polarized antigen-specific effector cells has been shown to be protective in mice challenged with a lethal dose of influenza, thus suggesting a protective role for IL-17 that is independent of IFN-γ137. In contrast, IL-17R-deficient mice have also been shown to have reduced neutrophil influx and decreased inflammation, suggesting a pathological role for IL-17 during influenza challenge138,139. The genetic background of mice used and the influenza dose used were different between the studies, suggesting a protective or pathological role for IL-17 in influenza. Therefore, these studies suggest that the genetic background and infectious dose may act as a determining factor regarding the protective or pathologic role of IL-17 during influenza infection. In contrast, IL-17 is associated with the pathology in 2009 pandemic influenza A (H1N1)-induced acute lung injury140. Additionally, IL-17 levels are associated with the exacerbated disease pathology induced following viral infections such as hepatitis141,142, vaccinia virus143,144, RSV145–147, HSV148,149, and EBV150,151. During viral infections (such as hepatitis), IL-17 can either potentiate early neutrophil infiltration at the site of infection152 or inhibit NK cell-mediated host immune response (for example, vaccinia virus infection)153. Neutralization of IL-17 not only reduced the disease severity but also reduced the viral load in the host and improved survival of the host during HSV72,148 and Dengue virus154 infections. Despite having a pathological role against most viral infections, IL-17 was suggested in several reports to have a protective role during HIV infection. Along with the Th17 cells, a subset of CD8+ cells which produce IL-17, also known as TC17, are important in the context of viral infection, although the detailed role of TC17 has yet to be delineated155,156. Moreover, Treg/Th17 ratios dictate the outcome of infection as well as effectiveness of anti-retroviral treatment157,158. Therefore, the balance between the Treg and Th17/Tc17 is suggested to be more important than that of the expression of IL-17 alone159. However, some recent data also suggest that during HIV infection IL-17 levels have a negative correlation with HIV plasma viral load160. Therefore, these data together suggest that IL-17 may be contributing to the inflammatory injury in response to viral infection, but the recruitment of inflammatory cells such as neutrophils or lymphocytes may be required for protection. We propose that the full array of IL-17 responses during various viral infections has yet to be fully delineated.
Anti-IL-17 therapies and impact on host immunity to infections
Exacerbated IL-17 production is linked to excessive inflammation-associated complications such as autoimmunity, chronic obstructive pulmonary disease (COPD), and contact dermatitis. Moreover, P. gingivalis infection predisposes the patient to the potential risk of acquiring autoimmune disorders, specifically rheumatoid arthritis (RA)161,162 through excessive inflammation (induced by IL-17) or generation of autoantibodies. As a result, diseases such as psoriasis163, RA164, and contact dermatitis165 are emerging as particularly strong IL-17-driven disorders. Similarly, excessive IL-17 leads to the upregulation of neutrophil-attracting chemokines and subsequent neutrophil infiltration and inflammation during COPD166,167. A number of biologic drugs targeting IL-17A/F and IL-17RA are being used or evaluated as treatment options against several diseases, such as COPD168, psoriasis, and RA, with impressive efficacy169,170. However, IL-17 is strongly associated with the protection against Mtb clinical isolates and fungal infections. IL-17 and IL-17RA single-nucleotide polymorphisms enhance the risk of fungal diseases such as candidiasis171 and bacterial disease such as pulmonary tuberculosis in certain cohorts172,173. Moreover, deficiency in CARD-9174 or gain of function of signal transducer and activator of transcription 1 (STAT-1)175 impairs IL-17 signaling and these mutations are associated with the chronic candidiasis. Therefore, we suggest that anti-IL-17 treatments may have a detrimental effect on the overall immunity of those individuals as they may become immunocompromised, resulting in predisposition toward the risk of acquiring several infections (including Candida176 and Mycobacterium177).
Conclusions
The importance of IL-17 in different infectious models is now well established. Although there are several infections where the role of IL-17 is not clear, IL-17 plays distinct yin-and-yang roles in a majority of the cases. IL-17 plays a protective role against the infection, and excess IL-17 promotes pathology and tissue destruction. The overall global role for the involvement of IL-17 in infection models is summarized in Figure 1 and Table 1. Upon exposure to pathogens (bacteria, fungus, or virus), myeloid cells produce factors that promote the production of IL-17 from both innate and adaptive cells. IL-17 then acts on primary responder cells (epithelial, macrophage, or myeloid cells), thereby inducing the production of other anti-microbial peptides, chemokines, and cytokines. IL-17-induced chemokines recruit neutrophils (and other immune cells) to the site of infection and restrict pathogenesis. On the other hand, this pathway can mediate excessive inflammation and exacerbated pathology at the infectious milieu. Hence, careful observation on the role of IL-17 is necessary to improve the overall treatment strategy against such infections. Therefore, it is important to critically consider the yin-and-yang roles of IL-17 while designing novel strategies to target specific pathways for control of pathogens.
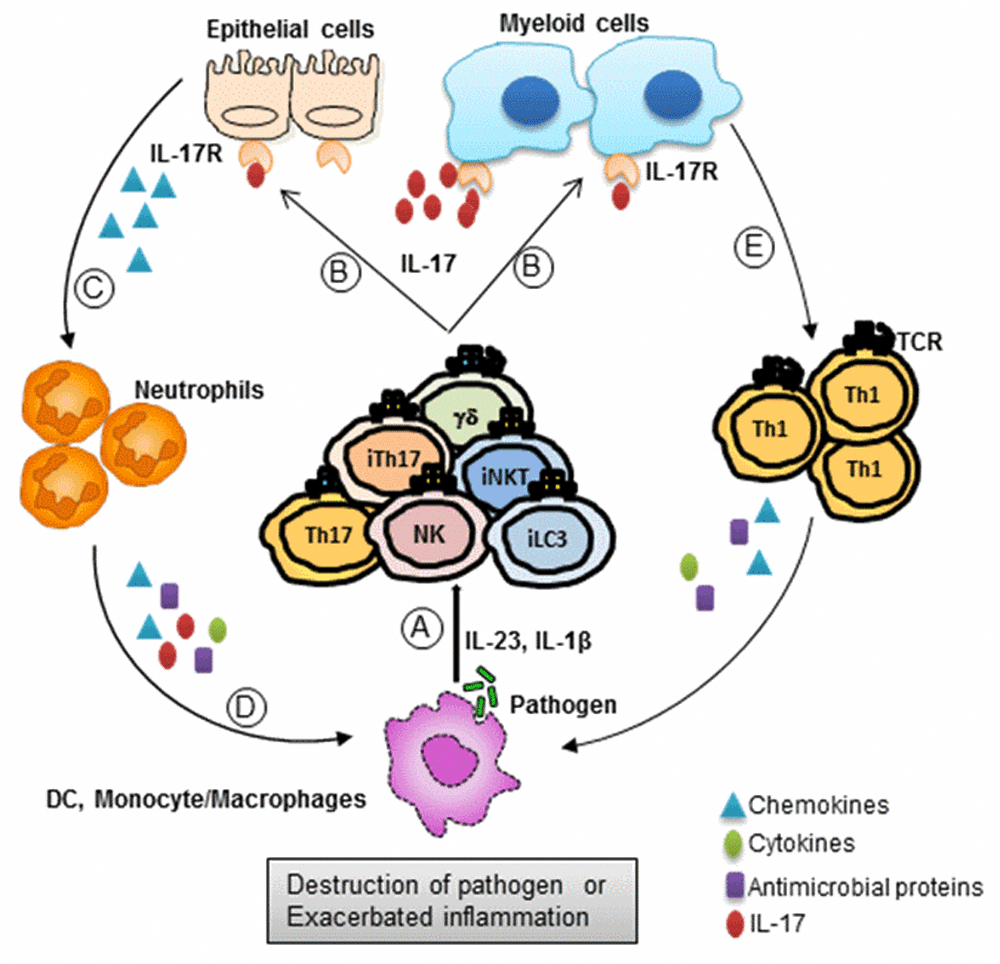
Figure 1. Yin-and-yang roles of IL-17 during infections.
As the host immune system encounters a pathogen, host immune cells respond by releasing an array of cytokines such as IL-23, IL-6, and IL-1β. (A) These cytokines elicit IL-17 production from both innate cells (iLC3, NK, iNKT, iTH17, and γδ T) and adaptive cells (Th17 and Tc17). (B) This IL-17 then acts on responder cells, which express IL-17Rs on the cell surface, such as epithelial cells or myeloid cells. (C) Through IL-17R signaling, these responder cells produce chemokines which help recruit neutrophils to the site of infection. (D) These recruited neutrophils destroy the pathogen (mostly extracellular) through the production of cytokines, chemokines, and anti-microbial peptides. (E) Similarly, myeloid cells are also able to restrict pathogen establishment through activation and recruitment of Th1 cells. These Th1 cells secrete proinflammatory cytokines, chemokines, and anti-microbial peptides to restrict pathogenesis. On the other hand, excessive inflammation at the site of infection may lead to exacerbated disease pathology. IL, interleukin; IL-17R, interleukin 17 receptor; iLC3, group 3 innate lymphoid cell; iNKT, invariant natural killer T; iTH17, innate T helper cell type 17 cell; NK, natural killer; Th, T helper cell type.
Table 1. Description of infections where protective or pathologic roles of IL-17 have been demonstrated.
| Protective roles of IL-17 | Pathologic role of IL-17 |
|---|
| Extracellular bacteria | Klebsiella pneumoniae48, Citrobacter rodentium49,
Staphylococcus aureus50, and Bordetella pertussis51 | Bordetella pertussis56,69,
Porphyromonas gingivalis66,
and Haemophilus influenza70 |
| Intracellular bacteria | Listeria monocytogenes71, Mycoplasma pulmonis72,
Legionella pneumophila73,74, Salmonella typhimurium75,
Chlamydia muridarum76, Francisella tularensis77, and
Mycobacterium tuberculosis78
| Mycobacterium tuberculosis96–98 |
| Parasites | Trypanosoma cruzi104, Toxoplasma gondii105,
Leishmania braziliensis106, and Echinococcus
granulosus107
| Leishmania major112,113,
Leishmania guyanensis114,
Eimeria tenella115, and
Toxoplasma gondii116 |
| Fungus | Candida albicans117, Cryptococcus neoformans118,
Pneumocystis carinii119, and Aspergillus fumigatus120 | Candida albicans130–133 and
Aspergillus fumigatus133 |
| Virus | H5N1134–136 and HIV154,155 | H1N1137–139, respiratory
syncytial virus144–146, herpes
simplex virus147,148, Epstein-Barr
virus149,150, vaccinia virus142,143,
Dengue virus153, hepatitis B and
C virus140,141, and HIV159
|
Abbreviations
CARD-9, caspase recruitment domain family member 9; COPD, chronic obstructive pulmonary disease; CXCL, C-X-C motif chemokine ligand; EBV, Epstein-Barr virus; G-CSF, granulocyte-colony-stimulating factor; GM-CSF, granulocyte macrophage-colony-stimulating factor; HIV, human immunodeficiency virus; HSV, herpes simplex virus; IFN-γ, interferon-gamma IL, interleukin; IL-17R, interleukin 17 receptor; iLC3, group 3 innate lymphoid cell; iNKT, invariant natural killer T; MIP-2, macrophage inflammatory protein 2; MyD88, myeloid differentiation factor 88; NF-κB, nuclear factor-kappa B; NK, natural killer; OEC, oral epithelial cell; PBMC, peripheral blood mononuclear cell; RA, rheumatoid arthritis; RSV, respiratory syncytial virus; TGF-β, transforming growth factor-beta; Th17, T helper cell type 17; TNF-α, tumor necrosis factor-alpha; TRAF-6, tumor necrosis factor receptor-associated factor 6; Treg, regulatory T cell.
Competing interests
The authors declare that they have no competing interests.
Grant information
This work was supported by Washington University in St. Louis and National Institutes of Health grants HL105427, AI111914, and AI123780.
Acknowledgments
We thank Kimberly Thomas for critical reading of the manuscript.
Faculty Opinions recommendedReferences
- 1.
Gu C, Wu L, Li X:
IL-17 family: cytokines, receptors and signaling.
Cytokine.
2013; 64(2): 477–85. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 2.
Kolls JK, Lindén A:
Interleukin-17 family members and inflammation.
Immunity.
2004; 21(4): 467–76. PubMed Abstract
| Publisher Full Text
- 3.
Iwakura Y, Ishigame H, Saijo S, et al.:
Functional specialization of interleukin-17 family members.
Immunity.
2011; 34(2): 149–62. PubMed Abstract
| Publisher Full Text
- 4.
Jin W, Dong C:
IL-17 cytokines in immunity and inflammation.
Emerg Microbes Infect.
2013; 2: e60. Publisher Full Text
- 5.
Gaffen SL:
Structure and signalling in the IL-17 receptor family.
Nat Rev Immunol.
2009; 9(8): 556–67. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 6.
Das Sarma J, Ciric B, Marek R, et al.:
Functional interleukin-17 receptor A is expressed in central nervous system glia and upregulated in experimental autoimmune encephalomyelitis.
J Neuroinflammation.
2009; 6: 14. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 7.
Ho AW, Gaffen SL:
IL-17RC: a partner in IL-17 signaling and beyond.
Semin Immunopathol.
2010; 32(1): 33–42. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 8.
Xie S, Li J, Wang JH, et al.:
IL-17 activates the canonical NF-kappaB signaling pathway in autoimmune B cells of BXD2 mice to upregulate the expression of regulators of G-protein signaling 16.
J Immunol.
2010; 184(5): 2289–96. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 9.
Gaffen SL, Jain R, Garg AV, et al.:
The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing.
Nat Rev Immunol.
2014; 14(9): 585–600. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 10.
Shen F, Gaffen SL:
Structure-function relationships in the IL-17 receptor: implications for signal transduction and therapy.
Cytokine.
2008; 41(2): 92–104. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 11.
Onishi RM, Gaffen SL:
Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease.
Immunology.
2010; 129(3): 311–21. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 12.
Qu F, Gao H, Zhu S, et al.:
TRAF6-dependent Act1 phosphorylation by the IκB kinase-related kinases suppresses interleukin-17-induced NF-κB activation.
Mol Cell Biol.
2012; 32(19): 3925–37. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 13.
Schwandner R, Yamaguchi K, Cao Z:
Requirement of tumor necrosis factor receptor-associated factor (TRAF)6 in interleukin 17 signal transduction.
J Exp Med.
2000; 191(7): 1233–40. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 14.
Wu L, Zepp J, Li X:
Function of Act1 in IL-17 family signaling and autoimmunity.
Adv Exp Med Biol.
2012; 946: 223–35. PubMed Abstract
| Publisher Full Text
- 15.
Chang SH, Park H, Dong C:
Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor.
J Biol Chem.
2006; 281(47): 35603–7. PubMed Abstract
| Publisher Full Text
- 16.
Kaiko GE, Horvat JC, Beagley KW, et al.:
Immunological decision-making: how does the immune system decide to mount a helper T-cell response?
Immunology.
2008; 123(3): 326–38. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 17.
Weaver CT, Harrington LE, Mangan PR, et al.:
Th17: an effector CD4 T cell lineage with regulatory T cell ties.
Immunity.
2006; 24(6): 677–88. PubMed Abstract
| Publisher Full Text
- 18.
Huehn J, Polansky JK, Hamann A:
Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage?
Nat Rev Immunol.
2009; 9(2): 83–9. PubMed Abstract
| Publisher Full Text
- 19.
Caza T, Landas S:
Functional and Phenotypic Plasticity of CD4+ T Cell Subsets.
Biomed Res Int.
2015; 2015: 521957. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 20.
Guglani L, Khader SA:
Th17 cytokines in mucosal immunity and inflammation.
Curr Opin HIV AIDS.
2010; 5(2): 120–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 21.
McGeachy MJ:
GM-CSF: the secret weapon in the TH17 arsenal.
Nat Immunol.
2011; 12(6): 521–2. PubMed Abstract
| Publisher Full Text
- 22.
Muranski P, Restifo NP:
Essentials of Th17 cell commitment and plasticity.
Blood.
2013; 121(13): 2402–14. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 23.
Evans CM, Jenner RG:
Transcription factor interplay in T helper cell differentiation.
Brief Funct Genomics.
2013; 12(6): 499–511. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 24.
Chien YH, Zeng X, Prinz I:
The natural and the inducible: interleukin (IL)-17-producing γδ T cells.
Trends Immunol.
2013; 34(4): 151–4. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 25.
Cortez VS, Robinette ML, Colonna M:
Innate lymphoid cells: new insights into function and development.
Curr Opin Immunol.
2015; 32: 71–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 26.
Ciccia F, Guggino G, Rizzo A, et al.:
Type 3 innate lymphoid cells producing IL-17 and IL-22 are expanded in the gut, in the peripheral blood, synovial fluid and bone marrow of patients with ankylosing spondylitis.
Ann Rheum Dis.
2015; 74(9): 1739–47. PubMed Abstract
| Publisher Full Text
- 27.
Taylor PR, Roy S, Leal SM Jr, et al.:
Activation of neutrophils by autocrine IL-17A-IL-17RC interactions during fungal infection is regulated by IL-6, IL-23, RORγt and dectin-2.
Nat Immunol.
2014; 15(2): 143–51. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 28.
Taylor PR, Leal SM Jr, Sun Y, et al.:
Aspergillus and Fusarium corneal infections are regulated by Th17 cells and IL-17-producing neutrophils.
J Immunol.
2014; 192(7): 3319–27. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 29.
Bi Y, Zhou J, Yang H, et al.:
IL-17A produced by neutrophils protects against pneumonic plague through orchestrating IFN-γ-activated macrophage programming.
J Immunol.
2014; 192(2): 704–13. PubMed Abstract
| Publisher Full Text
- 30.
Monteiro M, Almeida CF, Agua-Doce A, et al.:
Induced IL-17-producing invariant NKT cells require activation in presence of TGF-β and IL-1β.
J Immunol.
2013; 190(2): 805–11. PubMed Abstract
| Publisher Full Text
- 31.
Massot B, Michel M, Diem S, et al.:
TLR-induced cytokines promote effective proinflammatory natural Th17 cell responses.
J Immunol.
2014; 192(12): 5635–42. PubMed Abstract
| Publisher Full Text
- 32.
Passos ST, Silver JS, O'Hara AC, et al.:
IL-6 promotes NK cell production of IL-17 during toxoplasmosis.
J Immunol.
2010; 184(4): 1776–83. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 33.
Barthelemy A, Ivanov S, Hassane M, et al.:
Exogenous Activation of Invariant Natural Killer T Cells by α-Galactosylceramide Reduces Pneumococcal Outgrowth and Dissemination Postinfluenza.
mBio.
2016; 7(6): pii: e01440-16. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 34.
McCarthy MK, Zhu L, Procario MC, et al.:
IL-17 contributes to neutrophil recruitment but not to control of viral replication during acute mouse adenovirus type 1 respiratory infection.
Virology.
2014; 456–457: 259–67. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 35.
Lubberts E, Koenders MI, van den Berg WB:
The role of T-cell interleukin-17 in conducting destructive arthritis: lessons from animal models.
Arthritis Res Ther.
2005; 7(1): 29–37. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 36.
Dixon BR, Radin JN, Piazuelo MB, et al.:
IL-17a and IL-22 Induce Expression of Antimicrobials in Gastrointestinal Epithelial Cells and May Contribute to Epithelial Cell Defense against Helicobacter pylori.
PLoS One.
2016; 11(2): e0148514. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 37.
Wang CQ, Akalu YT, Suarez-Farinas M, et al.:
IL-17 and TNF synergistically modulate cytokine expression while suppressing melanogenesis: potential relevance to psoriasis.
J Invest Dermatol.
2013; 133(12): 2741–52. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 38.
Chiricozzi A, Nograles KE, Johnson-Huang LM, et al.:
IL-17 induces an expanded range of downstream genes in reconstituted human epidermis model.
PLoS One.
2014; 9(2): e90284. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 39.
Kolls JK, Khader SA:
The role of Th17 cytokines in primary mucosal immunity.
Cytokine Growth Factor Rev.
2010; 21(6): 443–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 40.
Ye P, Garvey PB, Zhang P, et al.:
Interleukin-17 and lung host defense against Klebsiella pneumoniae infection.
Am J Respir Cell Mol Biol.
2001; 25(3): 335–40. PubMed Abstract
| Publisher Full Text
- 41.
Hamada S, Umemura M, Shiono T, et al.:
IL-17A produced by gammadelta T cells plays a critical role in innate immunity against listeria monocytogenes infection in the liver.
J Immunol.
2008; 181(5): 3456–63. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 42.
Tenland E, Håkansson G, Alaridah N, et al.:
Innate Immune Responses after Airway Epithelial Stimulation with Mycobacterium bovis Bacille-Calmette Guérin.
PLoS One.
2016; 11(10): e0164431. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 43.
Broz P, Ohlson MB, Monack DM:
Innate immune response to Salmonella typhimurium, a model enteric pathogen.
Gut Microbes.
2012; 3(2): 62–70. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 44.
Gladiator A, LeibundGut-Landmann S:
Innate lymphoid cells: new players in IL-17-mediated antifungal immunity.
PLoS Pathog.
2013; 9(12): e1003763. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 45.
Martinez NE, Sato F, Kawai E, et al.:
Regulatory T cells and Th17 cells in viral infections: implications for multiple sclerosis and myocarditis.
Future Virol.
2012; 7(6): 593–608. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 46.
Saghaug CS, Sørnes S, Peirasmaki D, et al.:
Human Memory CD4+ T Cell Immune Responses against Giardia lamblia.
Clin Vaccine Immunol.
2015; 23(1): 11–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 47.
Suryawanshi A, Cao Z, Sampson JF, et al.:
IL-17A-mediated protection against Acanthamoeba keratitis.
J Immunol.
2015; 194(2): 650–63. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 48.
Ye P, Rodriguez FH, Kanaly S, et al.:
Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense.
J Exp Med.
2001; 194(4): 519–27. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 49.
Collins JW, Keeney KM, Crepin VF, et al.:
Citrobacter rodentium: infection, inflammation and the microbiota.
Nat Rev Microbiol.
2014; 12(9): 612–23. PubMed Abstract
| Publisher Full Text
- 50.
Murphy AG, O'Keeffe KM, Lalor SJ, et al.:
Staphylococcus aureus infection of mice expands a population of memory γδ T cells that are protective against subsequent infection.
J Immunol.
2014; 192(8): 3697–708. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 51.
Warfel JM, Merkel TJ:
Bordetella pertussis infection induces a mucosal IL-17 response and long-lived Th17 and Th1 immune memory cells in nonhuman primates.
Mucosal Immunol.
2013; 6(4): 787–96. PubMed Abstract
| Publisher Full Text
- 52.
Chan LC, Chaili S, Filler SG, et al.:
Nonredundant Roles of Interleukin-17A (IL-17A) and IL-22 in Murine Host Defense against Cutaneous and Hematogenous Infection Due to Methicillin-Resistant Staphylococcus aureus.
Infect Immun.
2015; 83(11): 4427–37. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 53.
Cheng P, Liu T, Zhou WY, et al.:
Role of gamma-delta T cells in host response against Staphylococcus aureus-induced pneumonia.
BMC Immunol.
2012; 13: 38. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 54.
Xiong H, Keith JW, Samilo DW, et al.:
Innate Lymphocyte/Ly6Chi Monocyte Crosstalk Promotes Klebsiella Pneumoniae Clearance.
Cell.
2016; 165(3): 679–89. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 55.
Price AE, Reinhardt RL, Liang HE, et al.:
Marking and quantifying IL-17A-producing cells in vivo.
PLoS One.
2012; 7(6): e39750. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 56.
Ross PJ, Sutton CE, Higgins S, et al.:
Relative contribution of Th1 and Th17 cells in adaptive immunity to Bordetella pertussis: towards the rational design of an improved acellular pertussis vaccine.
PLoS Pathog.
2013; 9(4): e1003264. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 57.
Chen K, McAleer JP, Lin Y, et al.:
Th17 cells mediate clade-specific, serotype-independent mucosal immunity.
Immunity.
2011; 35(6): 997–1009. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 58.
McAleer JP, Kolls JK:
Directing traffic: IL-17 and IL-22 coordinate pulmonary immune defense.
Immunol Rev.
2014; 260(1): 129–44. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 59.
Chen K, Eddens T, Trevejo-Nunez G, et al.:
IL-17 Receptor Signaling in the Lung Epithelium Is Required for Mucosal Chemokine Gradients and Pulmonary Host Defense against K. pneumoniae.
Cell Host Microbe.
2016; 20(5): 596–605. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 60.
Brown AF, Murphy AG, Lalor SJ, et al.:
Memory Th1 Cells Are Protective in Invasive Staphylococcus aureus Infection.
PLoS Pathog.
2015; 11(11): e1005226. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 61.
Ishigame H, Kakuta S, Nagai T, et al.:
Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses.
Immunity.
2009; 30(1): 108–19. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 62.
Prabhakara R, Foreman O, De Pascalis R, et al.:
Epicutaneous model of community-acquired Staphylococcus aureus skin infections.
Infect Immun.
2013; 81(4): 1306–15. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 63.
Witowski J, Pawlaczyk K, Breborowicz A, et al.:
IL-17 stimulates intraperitoneal neutrophil infiltration through the release of GRO alpha chemokine from mesothelial cells.
J Immunol.
2000; 165(10): 5814–21. PubMed Abstract
| Publisher Full Text
- 64.
Sutton CE, Mielke LA, Mills KH:
IL-17-producing γδ T cells and innate lymphoid cells.
Eur J Immunol.
2012; 42(9): 2221–31. PubMed Abstract
| Publisher Full Text
- 65.
Cai S, Batra S, Del Piero F, et al.:
NLRP12 modulates host defense through IL-17A-CXCL1 axis.
Mucosal Immunol.
2016; 9(2): 503–14. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 66.
de Aquino SG, Abdollahi-Roodsaz S, Koenders MI, et al.:
Periodontal pathogens directly promote autoimmune experimental arthritis by inducing a TLR2- and IL-1-driven Th17 response.
J Immunol.
2014; 192(9): 4103–11. PubMed Abstract
| Publisher Full Text
- 67.
Corrêa JD, Madeira MF, Resende RG, et al.:
Association between polymorphisms in interleukin-17A and -17F genes and chronic periodontal disease.
Mediators Inflamm.
2012; 2012: 846052. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 68.
Cheng W, van Asten SD, Burns LA, et al.:
Periodontitis-associated pathogens P. gingivalis and A. actinomycetemcomitans activate human CD14+ monocytes leading to enhanced Th17/IL-17 responses.
Eur J Immunol.
2016; 46(9): 2211–21. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 69.
Connelly CE, Sun Y, Carbonetti NH:
Pertussis toxin exacerbates and prolongs airway inflammatory responses during Bordetella pertussis infection.
Infect Immun.
2012; 80(12): 4317–32. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 70.
Essilfie AT, Simpson JL, Dunkley ML, et al.:
Combined Haemophilus influenzae respiratory infection and allergic airways disease drives chronic infection and features of neutrophilic asthma.
Thorax.
2012; 67(7): 588–99. PubMed Abstract
| Publisher Full Text
- 71.
Sheridan BS, Romagnoli PA, Pham QM, et al.:
γδ T cells exhibit multifunctional and protective memory in intestinal tissues.
Immunity.
2013; 39(1): 184–95. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 72.
Way EE, Chen K, Kolls JK:
Dysregulation in lung immunity - the protective and pathologic Th17 response in infection.
Eur J Immunol.
2013; 43(12): 3116–24. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 73.
Kimizuka Y, Kimura S, Saga T, et al.:
Roles of interleukin-17 in an experimental Legionella pneumophila pneumonia model.
Infect Immun.
2012; 80(3): 1121–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 74.
Cai S, Batra S, Langohr I, et al.:
IFN-γ induction by neutrophil-derived IL-17A homodimer augments pulmonary antibacterial defense.
Mucosal Immunol.
2016; 9(3): 718–29. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 75.
Keestra AM, Godinez I, Xavier MN, et al.:
Early MyD88-dependent induction of interleukin-17A expression during Salmonella colitis.
Infect Immun.
2011; 79(8): 3131–40. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 76.
O'Meara CP, Armitage CW, Harvie MC, et al.:
Immunity against a Chlamydia infection and disease may be determined by a balance of IL-17 signaling.
Immunol Cell Biol.
2014; 92(3): 287–97. PubMed Abstract
| Publisher Full Text
- 77.
Skyberg JA, Rollins MF, Samuel JW, et al.:
Interleukin-17 protects against the Francisella tularensis live vaccine strain but not against a virulent F. tularensis type A strain.
Infect Immun.
2013; 81(9): 3099–105. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 78.
Monin L, Griffiths KL, Slight S, et al.:
Immune requirements for protective Th17 recall responses to Mycobacterium tuberculosis challenge.
Mucosal Immunol.
2015; 8(5): 1099–109. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 79.
Gao Y, Williams AP:
Role of Innate T Cells in Anti-Bacterial Immunity.
Front Immuno.l.
2015; 6: 302. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 80.
Lyadova IV, Panteleev AV:
Th1 and Th17 Cells in Tuberculosis: Protection, Pathology, and Biomarkers.
Mediators Inflamm.
2015; 2015: 854507. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 81.
Umemura M, Okamoto-Yoshida Y, Yahagi A, et al.:
Involvement of IL-17A-producing TCR γδ T cells in late protective immunity against pulmonary Mycobacterium tuberculosis infection.
Immun Inflamm Dis.
2016; 4(4): 401–12. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 82.
Henry T, Kirimanjeswara GS, Ruby T, et al.:
Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections.
J Immunol.
2010; 184(7): 3755–67. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 83.
Steinbach S, Vordermeier HM, Jones GJ:
CD4+ and γδ T Cells are the main Producers of IL-22 and IL-17A in Lymphocytes from Mycobacterium bovis-infected Cattle.
Sci Rep.
2016; 6: 29990. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 84.
Pick J, Arra A, Lingel H, et al.:
CTLA-4 (CD152) enhances the Tc17 differentiation program.
Eur J Immunol.
2014; 44(7): 2139–52. PubMed Abstract
| Publisher Full Text
- 85.
Behnsen J, Perez-Lopez A, Nuccio SP, et al.:
Exploiting host immunity: the Salmonella paradigm.
Trends Immunol.
2015; 36(2): 112–20. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 86.
Freches D, Korf H, Denis O, et al.:
Mice genetically inactivated in interleukin-17A receptor are defective in long-term control of Mycobacterium tuberculosis infection.
Immunology.
2013; 140(2): 220–31. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 87.
Orgun NN, Mathis MA, Wilson CB, et al.:
Deviation from a strong Th1-dominated to a modest Th17-dominated CD4 T cell response in the absence of IL-12p40 and type I IFNs sustains protective CD8 T cells.
J Immunol.
2008; 180(6): 4109–15. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 88.
Zhang X, Gao L, Lei L, et al.:
A MyD88-dependent early IL-17 production protects mice against airway infection with the obligate intracellular pathogen Chlamydia muridarum.
J Immunol.
2009; 183(2): 1291–300. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 89.
Slight SR, Monin L, Gopal R, et al.:
IL-10 restrains IL-17 to limit lung pathology characteristics following pulmonary infection with Francisella tularensis live vaccine strain.
Am J Pathol.
2013; 183(5): 1397–404. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 90.
Khader SA, Pearl JE, Sakamoto K, et al.:
IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available.
J Immunol.
2005; 175(2): 788–95. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 91.
Segueni N, Tritto E, Bourigault ML, et al.:
Controlled Mycobacterium tuberculosis infection in mice under treatment with anti-IL-17A or IL-17F antibodies, in contrast to TNFα neutralization.
Sci Rep.
2016; 6: 36923. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 92.
Khader SA, Gopal R:
IL-17 in protective immunity to intracellular pathogens.
Virulence.
2010; 1(5): 423–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 93.
Gopal R, Rangel-Moreno J, Slight S, et al.:
Interleukin-17-dependent CXCL13 mediates mucosal vaccine-induced immunity against tuberculosis.
Mucosal Immunol.
2013; 6(5): 972–84. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 94.
Khader SA, Bell GK, Pearl JE, et al.:
IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge.
Nat Immunol.
2007; 8(4): 369–77. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 95.
Gopal R, Monin L, Slight S, et al.:
Unexpected role for IL-17 in protective immunity against hypervirulent Mycobacterium tuberculosis HN878 infection.
PLoS Pathog.
2014; 10(5): e1004099. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 96.
Li Q, Li J, Tian J, et al.:
IL-17 and IFN-γ production in peripheral blood following BCG vaccination and Mycobacterium tuberculosis infection in human.
Eur Rev Med Pharmacol Sci.
2012; 16(14): 2029–36. PubMed Abstract
- 97.
Basile JI, Geffner LJ, Romero MM, et al.:
Outbreaks of mycobacterium tuberculosis MDR strains induce high IL-17 T-cell response in patients with MDR tuberculosis that is closely associated with high antigen load.
J Infect Dis.
2011; 204(7): 1054–64. PubMed Abstract
| Publisher Full Text
- 98.
Cruz A, Fraga AG, Fountain JJ, et al.:
Pathological role of interleukin 17 in mice subjected to repeated BCG vaccination after infection with Mycobacterium tuberculosis.
J Exp Med.
2010; 207(8): 1609–16. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 99.
Gopal R, Monin L, Torres D, et al.:
S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis.
Am J Respir Crit Care Med.
2013; 188(9): 1137–46. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 100.
Flierl MA, Rittirsch D, Gao H, et al.:
Adverse functions of IL-17A in experimental sepsis.
FASEB J.
2008; 22(7): 2198–205. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 101.
Ogiku M, Kono H, Hara M, et al.:
Interleukin-17A plays a pivotal role in polymicrobial sepsis according to studies using IL-17A knockout mice.
J Surg Res.
2012; 174(1): 142–9. PubMed Abstract
| Publisher Full Text
- 102.
Luo CJ, Luo F, Zhang L, et al.:
Knockout of interleukin-17A protects against sepsis-associated acute kidney injury.
Ann Intensive Care.
2016; 6(1): 56. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 103.
Wynn JL, Wilson CS, Hawiger J, et al.:
Targeting IL-17A attenuates neonatal sepsis mortality induced by IL-18.
Proc Natl Acad Sci U S A.
2016; 113(19): E2627–35. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 104.
Wu HP, Shih CC, Chu CM, et al.:
Effect of interleukin-17 on in vitro cytokine production in healthy controls and patients with severe sepsis.
J Formos Med Assoc.
2015; 114(12): 1250–7. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 105.
Erdmann H, Roßnagel C, Böhme J, et al.:
IL-17A promotes macrophage effector mechanisms against Trypanosoma cruzi by trapping parasites in the endolysosomal compartment.
Immunobiology.
2013; 218(6): 910–23. PubMed Abstract
| Publisher Full Text
- 106.
Peckham RK, Brill R, Foster DS, et al.:
Two distinct populations of bovine IL-17+ T-cells can be induced and WC1+IL-17+γδ T-cells are effective killers of protozoan parasites.
Sci Rep.
2014; 4: 5431. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 107.
de Oliveira CI, Brodskyn CI:
The immunobiology of Leishmania braziliensis infection.
Front Immunol.
2012; 3: 145. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 108.
Mezioug D, Touil-Boukoffa C:
Interleukin-17A correlates with interleukin-6 production in human cystic echinococcosis: a possible involvement of IL-17A in immunoprotection against Echinococcus granulosus infection.
Eur Cytokine Netw.
2012; 23(3): 112–9. PubMed Abstract
| Publisher Full Text
- 109.
Silva JL, Rezende-Oliveira K, da Silva MV, et al.:
IL-17-expressing CD4+ and CD8+ T lymphocytes in human toxoplasmosis.
Mediators Inflamm.
2014; 2014: 573825. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 110.
Novoa R, Bacellar O, Nascimento M, et al.:
IL-17 and Regulatory Cytokines (IL-10 and IL-27) in L. braziliensis Infection.
Parasite Immunol.
2011; 33(2): 132–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 111.
Tosello Boari J, Amezcua Vesely MC, Bermejo DA, et al.:
IL-17RA signaling reduces inflammation and mortality during Trypanosoma cruzi infection by recruiting suppressive IL-10-producing neutrophils.
PLoS Pathog.
2012; 8(4): e1002658. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 112.
Pang N, Zhang F, Ma X, et al.:
TGF-β/Smad signaling pathway regulates Th17/Treg balance during Echinococcus multilocularis infection.
Int Immunopharmacol.
2014; 20(1): 248–57. PubMed Abstract
| Publisher Full Text
- 113.
Gonzalez-Lombana C, Gimblet C, Bacellar O, et al.:
IL-17 mediates immunopathology in the absence of IL-10 following Leishmania major infection.
PLoS Pathog.
2013; 9(3): e1003243. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 114.
Boaventura VS, Santos CS, Cardoso CR, et al.:
Human mucosal leishmaniasis: neutrophils infiltrate areas of tissue damage that express high levels of Th17-related cytokines.
Eur J Immunol.
2010; 40(10): 2830–6. PubMed Abstract
| Publisher Full Text
- 115.
Hartley MA, Bourreau E, Rossi M, et al.:
Leishmaniavirus-Dependent Metastatic Leishmaniasis Is Prevented by Blocking IL-17A.
PLoS Pathog.
2016; 12(9): e1005852. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 116.
Del Cacho E, Gallego M, Lillehoj HS, et al.:
IL-17A regulates Eimeria tenella schizont maturation and migration in avian coccidiosis.
Vet Res.
2014; 45: 25. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 117.
Guiton R, Vasseur V, Charron S, et al.:
Interleukin 17 receptor signaling is deleterious during Toxoplasma gondii infection in susceptible BL6 mice.
J Infect Dis.
2010; 202(3): 427–35. PubMed Abstract
| Publisher Full Text
- 118.
Trautwein-Weidner K, Gladiator A, Nur S, et al.:
IL-17-mediated antifungal defense in the oral mucosa is independent of neutrophils.
Mucosal Immunol.
2015; 8(2): 221–31. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 119.
Murdock BJ, Huffnagle GB, Olszewski MA, et al.:
Interleukin-17A enhances host defense against cryptococcal lung infection through effects mediated by leukocyte recruitment, activation, and gamma interferon production.
Infect Immun.
2014; 82(3): 937–48. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 120.
Rudner XL, Happel KI, Young EA, et al.:
Interleukin-23 (IL-23)-IL-17 cytokine axis in murine Pneumocystis carinii infection.
Infect Immun.
2007; 75(6): 3055–61. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 121.
Guerra ES, Lee CK, Specht CA, et al.:
Central Role of IL-23 and IL-17 Producing Eosinophils as Immunomodulatory Effector Cells in Acute Pulmonary Aspergillosis and Allergic Asthma.
PLoS Pathog.
2017; 13(1): e1006175. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 122.
Sparber F, LeibundGut-Landmann S:
Interleukin 17-Mediated Host Defense against Candida albicans.
Pathogens.
2015; 4(3): 606–19. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 123.
Gladiator A, Wangler N, Trautwein-Weidner K, et al.:
Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection.
J Immunol.
2013; 190(2): 521–5. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 124.
Bär E, Whitney PG, Moor K, et al.:
IL-17 regulates systemic fungal immunity by controlling the functional competence of NK cells.
Immunity.
2014; 40(1): 117–27. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 125.
Ravikumar S, Win MS, Chai LY:
Optimizing Outcomes in Immunocompromised Hosts: Understanding the Role of Immunotherapy in Invasive Fungal Diseases.
Front Microbiol.
2015; 6: 1322. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 126.
Conti HR, Bruno VM, Childs EE, et al.:
IL-17 Receptor Signaling in Oral Epithelial Cells Is Critical for Protection against Oropharyngeal Candidiasis.
Cell Host Microbe.
2016; 20(5): 606–17. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 127.
Drummond RA, Collar AL, Swamydas M, et al.:
CARD9-Dependent Neutrophil Recruitment Protects against Fungal Invasion of the Central Nervous System.
PLoS Pathog.
2015; 11(12): e1005293. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 128.
Drewniak A, Gazendam RP, Tool AT, et al.:
Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency.
Blood.
2013; 121(13): 2385–92. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 129.
Huppler AR, Bishu S, Gaffen SL:
Mucocutaneous candidiasis: the IL-17 pathway and implications for targeted immunotherapy.
Arthritis Res Ther.
2012; 14(4): 217. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 130.
Ouederni M, Sanal O, Ikinciogullari A, et al.:
Clinical features of Candidiasis in patients with inherited interleukin 12 receptor β1 deficiency.
Clin Infect Dis.
2014; 58(2): 204–13. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 131.
Huang J, Meng S, Hong S, et al.:
IL-17C is required for lethal inflammation during systemic fungal infection.
Cell Mol Immunol.
2016; 13(4): 474–83. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 132.
Zelante T, De Luca A, Bonifazi P, et al.:
IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance.
Eur J Immunol.
2007; 37(10): 2695–706. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 133.
De Luca A, Zelante T, D'Angelo C, et al.:
IL-22 defines a novel immune pathway of antifungal resistance.
Mucosal Immunol.
2010; 3(4): 361–73. PubMed Abstract
| Publisher Full Text
- 134.
Werner JL, Gessner MA, Lilly LM, et al.:
Neutrophils produce interleukin 17A (IL-17A) in a dectin-1- and IL-23-dependent manner during invasive fungal infection.
Infect Immun.
2011; 79(10): 3966–77. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 135.
Wang X, Chan CC, Yang M, et al.:
A critical role of IL-17 in modulating the B-cell response during H5N1 influenza virus infection.
Cell Mol Immunol.
2011; 8(6): 462–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 136.
Hamada H, Garcia-Hernandez Mde L, Reome JB, et al.:
Tc17, a unique subset of CD8 T cells that can protect against lethal influenza challenge.
J Immunol.
2009; 182(6): 3469–81. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 137.
McKinstry KK, Strutt TM, Buck A, et al.:
IL-10 deficiency unleashes an influenza-specific Th17 response and enhances survival against high-dose challenge.
J Immunol.
2009; 182(12): 7353–63. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 138.
Crowe CR, Chen K, Pociask DA, et al.:
Critical role of IL-17RA in immunopathology of influenza infection.
J Immunol.
2009; 183(8): 5301–10. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 139.
Gopal R, Rangel-Moreno J, Fallert Junecko BA, et al.:
Mucosal pre-exposure to Th17-inducing adjuvants exacerbates pathology after influenza infection.
Am J Pathol.
2014; 184(1): 55–63. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 140.
Li C, Yang P, Sun Y, et al.:
IL-17 response mediates acute lung injury induced by the 2009 pandemic influenza A (H1N1) virus.
Cell Res.
2012; 22(3): 528–38. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 141.
Bălănescu P, Lădaru A, Voiosu T, et al.:
Th17 and IL-17 immunity in chronic hepatitis C infection.
Rom J Intern Med.
2012; 50(1): 13–8. PubMed Abstract
- 142.
Macek Jilkova Z, Afzal S, Marche H, et al.:
Progression of fibrosis in patients with chronic viral hepatitis is associated with IL-17+ neutrophils.
Liver Int.
2016; 36(8): 1116–24. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 143.
Darling AR, Freyschmidt EJ, Burton OT:
IL-10 suppresses IL-17-mediated dermal inflammation and reduces the systemic burden of Vaccinia virus in a mouse model of eczema vaccinatum.
Clin Immunol.
2014; 150(2): 153–60. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 144.
Cush SS, Reynoso GV, Kamenyeva O, et al.:
Locally Produced IL-10 Limits Cutaneous Vaccinia Virus Spread.
PLoS Pathog.
2016; 12(3): e1005493. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 145.
Mukherjee S, Lindell DM, Berlin AA, et al.:
IL-17-induced pulmonary pathogenesis during respiratory viral infection and exacerbation of allergic disease.
Am J Pathol.
2011; 179(1): 248–58. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 146.
de Almeida Nagata DE, Demoor T, Ptaschinski C, et al.:
IL-27R-mediated regulation of IL-17 controls the development of respiratory syncytial virus-associated pathogenesis.
Am J Pathol.
2014; 184(6): 1807–18. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 147.
Stoppelenburg AJ, Salimi V, Hennus M, et al.:
Local IL-17A potentiates early neutrophil recruitment to the respiratory tract during severe RSV infection.
PLoS One.
2013; 8(10): e78461. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 148.
Suryawanshi A, Veiga-Parga T, Rajasagi NK, et al.:
Role of IL-17 and Th17 cells in herpes simplex virus-induced corneal immunopathology.
J Immunol.
2011; 187(4): 1919–30. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 149.
Rolinski J, Hus I:
Immunological aspects of acute and recurrent herpes simplex keratitis.
J Immunol Res.
2014; 2014: 513560. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 150.
Rahal EA, Hajjar H, Rajeh M:
Epstein-Barr Virus and Human herpes virus 6 Type A DNA Enhance IL-17 Production in Mice.
Viral Immunol.
2015; 28(5): 297–302. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 151.
Ohta R, Imai M, Kawada J, et al.:
Interleukin-17A-producing T lymphocytes in chronic active Epstein-Barr virus infection.
Microbiol Immunol.
2013; 57(2): 139–44. PubMed Abstract
| Publisher Full Text
- 152.
Savarin C, Stohlman SA, Hinton DR, et al.:
IFN-γ protects from lethal IL-17 mediated viral encephalomyelitis independent of neutrophils.
J Neuroinflammation.
2012; 9: 104. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 153.
Kawakami Y, Tomimori Y, Yumoto K, et al.:
Inhibition of NK cell activity by IL-17 allows vaccinia virus to induce severe skin lesions in a mouse model of eczema vaccinatum.
J Exp Med.
2009; 206(6): 1219–25. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 154.
Guabiraba R, Besnard A, Marques RE, et al.:
IL-22 modulates IL-17A production and controls inflammation and tissue damage in experimental dengue infection.
Eur J Immunol.
2013; 43(6): 1529–44. PubMed Abstract
| Publisher Full Text
- 155.
d'Ettorre G, Ceccarelli G, Andreotti M, et al.:
Analysis of Th17 and Tc17 Frequencies and Antiviral Defenses in Gut-Associated Lymphoid Tissue of Chronic HIV-1 Positive Patients.
Mediators Inflamm.
2015; 2015: 395484. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 156.
Gaardbo JC, Hartling HJ, Thorsteinsson K, et al.:
CD3+CD8+CD161high Tc17 cells are depleted in HIV-infection.
AIDS.
2013; 27(4): 659–62. PubMed Abstract
| Publisher Full Text
- 157.
Falivene J, Ghiglione Y, Laufer N, et al.:
Th17 and Th17/Treg ratio at early HIV infection associate with protective HIV-specific CD8+ T-cell responses and disease progression.
Sci Rep.
2015; 5: 11511. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 158.
Valverde-Villegas JM, Matte MC, de Medeiros RM, et al.:
New Insights about Treg and Th17 Cells in HIV Infection and Disease Progression.
J Immunol Res.
2015; 2015: 647916. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 159.
Nigam P, Kwa S, Velu V, et al.:
Loss of IL-17-producing CD8 T cells during late chronic stage of pathogenic simian immunodeficiency virus infection.
J Immunol.
2011; 186(2): 745–53. PubMed Abstract
| Publisher Full Text
- 160.
Singh A, Vajpayee M, Ali SA, et al.:
Loss of RORγt DNA binding activity inhibits IL-17 expression in HIV-1 infected Indian individuals.
Viral Immunol.
2013; 26(1): 60–7. PubMed Abstract
| Publisher Full Text
- 161.
Cheng WC, Hughes FJ, Taams LS:
The presence, function and regulation of IL-17 and Th17 cells in periodontitis.
J Clin Periodontol.
2014; 41(6): 541–9. PubMed Abstract
| Publisher Full Text
- 162.
Arunachalam LT:
Autoimmune correlation of rheumatoid arthritis and periodontitis.
J Indian Soc Periodontol.
2014; 18(5): 666–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 163.
Muromoto R, Hirao T, Tawa K, et al.:
IL-17A plays a central role in the expression of psoriasis signature genes through the induction of IκB-ζ in keratinocytes.
Int Immunol.
2016; 28(9): 443–52. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 164.
Lubberts E:
The IL-23-IL-17 axis in inflammatory arthritis.
Nat Rev Rheumatol.
2015; 11(10): 562. PubMed Abstract
| Publisher Full Text
- 165.
Peiser M:
Role of Th17 cells in skin inflammation of allergic contact dermatitis.
Clin Dev Immunol.
2013; 2013: 261037. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 166.
Cazzola M, Matera MG:
IL-17 in chronic obstructive pulmonary disease.
Expert Rev Respir Med.
2012; 6(2): 135–8. PubMed Abstract
| Publisher Full Text
- 167.
Roos AB, Sanden C, Mori M, et al.:
IL-17A Is Elevated in End-Stage Chronic Obstructive Pulmonary Disease and Contributes to Cigarette Smoke-induced Lymphoid Neogenesis.
Am J Respir Crit Care Med.
2015; 191(11): 1232–41. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 168.
Barnes PJ:
New anti-inflammatory targets for chronic obstructive pulmonary disease.
Nat Rev Drug Discov.
2013; 12(7): 543–59. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 169.
Kunwar S, Dahal K, Sharma S:
Anti-IL-17 therapy in treatment of rheumatoid arthritis: a systematic literature review and meta-analysis of randomized controlled trials.
Rheumatol Int.
2016; 36(8): 1065–75. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 170.
Canavan TN, Elmets CA, Cantrell WL, et al.:
Anti-IL-17 Medications Used in the Treatment of Plaque Psoriasis and Psoriatic Arthritis: A Comprehensive Review.
Am J Clin Dermatol.
2016; 17(1): 33–47. PubMed Abstract
| Publisher Full Text
- 171.
Plantinga TS, Johnson MD, Scott WK, et al.:
Human genetic susceptibility to Candida infections.
Med Mycol.
2012; 50(8): 785–94. PubMed Abstract
| Publisher Full Text
- 172.
Wang M, Xu G, Lü L, et al.:
Genetic polymorphisms of IL-17A, IL-17F, TLR4 and miR-146a in association with the risk of pulmonary tuberculosis.
Sci Rep.
2016; 6: 28586. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 173.
Rolandelli A, Hernández Del Pino RE, Pellegrini JM, et al.:
The IL-17A rs2275913 single nucleotide polymorphism is associated with protection to tuberculosis but related to higher disease severity in Argentina.
Sci Rep.
2017; 7: 40666. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 174.
Bishu S, Hernández-Santos N, Simpson-Abelson MR, et al.:
The adaptor CARD9 is required for adaptive but not innate immunity to oral mucosal Candida albicans infections.
Infect Immun.
2014; 82(3): 1173–80. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 175.
Yamazaki Y, Yamada M, Kawai T, et al.:
Two novel gain-of-function mutations of STAT1 responsible for chronic mucocutaneous candidiasis disease: impaired production of IL-17A and IL-22, and the presence of anti-IL-17F autoantibody.
J Immunol.
2014; 193(10): 4880–7. PubMed Abstract
| Publisher Full Text
- 176.
Langley RG, Elewski BE, Lebwohl M, et al.:
Secukinumab in plaque psoriasis--results of two phase 3 trials.
N Engl J Med.
2014; 371(4): 326–38. PubMed Abstract
| Publisher Full Text
- 177.
Balato N, Di Costanzo L, Ayala F, et al.:
Psoriatic disease and tuberculosis nowadays.
Clin Dev Immunol.
2012; 2012: 747204. PubMed Abstract
| Publisher Full Text
| Free Full Text
Comments on this article Comments (0)