1Center for Systems Biology, Lunenfeld-Tanenbaum Research Institute, Mount Sinai Hospital, Toronto, ON, Canada 2Department of Biochemistry and Donnelly Centre, University of Toronto, Toronto, ON, Canada 3Department of Molecular Genetics, University of Toronto, Toronto, ON, Canada
OPEN PEER REVIEW
REVIEWER STATUS
Abstract
The appearance of the first animal species on earth coincides with the emergence of transforming growth factor β (TGFβ) pathways. The evolution of these animals into more complex organisms coincides with a progressively increased TGFβ repertoire through gene duplications and divergence, making secreted TGFβ molecules the largest family of morphogenetic proteins in humans. It is therefore not surprising that TGFβ pathways govern numerous aspects of human biology from early embryonic development to regeneration, hematopoiesis, neurogenesis, and immunity. Such heavy reliance on these pathways is reflected in the susceptibility to minor perturbations in pathway components that can lead to dysregulated signaling and a diverse range of human pathologies such as cancer, fibrosis, and developmental disorders. Attempts to comprehensively resolve these signaling cascades are complicated by the long-recognized paradoxical role the pathway plays in cell biology. Recently, several groups have probed examples of the disparate aspects of TGFβ biology in a variety of animal models and uncovered novel context-dependent regulatory mechanisms. Here, we briefly review recent advancements and discuss their overall impact in directing future TGFβ research.
Corresponding authors:
Liliana Attisano, Jeffrey L Wrana
Competing interests:
The authors declare that they have no competing interests.
Grant information:
Work from the authors’ labs is supported by grants from the Canadian Institutes of Health Research (CIHR) Foundation program, the Terry Fox Research Institute, and the Krembil Foundation to LA and JLW. AA is also supported by a postdoctoral fellow award, part of the University of Toronto’s Medicine by Design initiative, which receives funding from the Canada First Research Excellence Fund (CFREF).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Transforming growth factor β (TGFβ) signaling pathways have co-evolved with animals1. Ligands of the TGFβ superfamily such as TGFβ(1–3), bone morphogenetic proteins (BMPs), activins, nodal, and growth differentiation factors (GDFs) control numerous aspects of animal biology including embryonic development, organogenesis, cell fate decisions, immune modulation, stress responses, and stem cell function2–7. Owing to widespread functional diversity, disruptions in TGFβ signaling are also associated with human diseases including cancer, fibrosis, systemic sclerosis, and hereditary disorders8–12. TGFβ morphogens often drive contradictory biological outcomes (Figure 1), which has perplexed investigators for decades2,9,13–15. For example, TGFβ1, first described as a transforming growth factor, is also a potent growth inhibitor at benign stages of cancer and promotes metastasis, genetic heterogeneity, and drug resistance in aggressive carcinomas8,9,13,14. Similarly, BMPs also have diverse functions. BMPs prevent self-renewal of the intestinal stem cells (ISCs) by inhibiting Wingless-type MMTV integration site (WNT)/β-catenin signaling, yet promote specification, expansion, and homing of hematopoietic progenitors16–21. Although multi-layer regulation of TGFβ signaling can theoretically explain the dichotomy of activities, the exact molecular contexts that lead to opposite biological output in intact tissues remain elusive. Recent studies utilizing multiple animal and human models have addressed these contradictions and uncovered complex interactions in immune and regenerative tissues that determine specific biological outcomes of TGFβ activity. This review will briefly summarize these findings.
Diverse biological activities regulated by TGFβ pathways are rigorously controlled in a finely calibrated contextual framework. Perturbation in these processes can lead to disease development.
TGFβ signal transduction
Canonical TGFβ Smad signaling is fairly simple2,6,22. Activated ligands, released by the cleavage of their pro-domains in the extracellular matrix, bind to a complex of two type II and type I transmembrane receptors, inducing type II-mediated phosphorylation of type I receptors at specific serine/threonine residues. This in turn triggers the activation of regulatory Smad (R-Smad) transcription factors, which then complex with the co-transcription factor Smad4 and accumulate in the nucleus, where they alter gene expression2,6,22. Inhibitory Smads (I-Smads), which are themselves induced by TGFβ family signaling23, function in a negative feedback loop to regulate TGFβ signaling. The mammalian genome encodes 33 ligands, five type II and seven type I receptors, and eight Smad proteins (including five R-Smads, two I-Smads, and a single co-Smad, Smad4). In Drosophila, there are seven ligands, three type II receptors, two type I receptors, two R-Smads (Mad and dSmad2, called Smox), one co-Smad (Medea or Med), and one I-Smad (Dad)6,22,24,25. TGFβ signaling is further divided into discrete pathways, as functionally redundant TGFβ ligands generally activate a restricted set of corresponding receptors and Smad transcription factors. For example, TGFβ(1–3) activate Smad2/3 via TGFβ receptors, BMPs stimulate Smad1/5/8 activation via BMP or activin receptors, and activins, inhibins, and GDFs activate activin receptors to phosphorylate Smad2/32,6,22. However, there are exceptions to this general model, as ligand/receptor promiscuity can lead to crosstalk between these distinct pathways26,27.
TGFβ signaling in intestinal repair and colon cancer
Tissue homeostasis is maintained by a rapidly cycling population of LGR5+ crypt base columnar ISCs that reside at the bottom of crypts of Lieberkühn28. As ISCs divide asymmetrically, their progeny expands in the transit-amplifying compartment and differentiates into functional lineages as they migrate towards the tips of villi. This baso-apical structural morphology in the mammalian intestine is maintained by two opposing concentration gradients of WNT and BMP signaling pathways29 that sustain the basal regenerative stem/progenitor cell zone and apical differentiated zone, respectively. Indeed, in humans, inactivating mutations in adenomatous polyposis coli (APC) as well as the BMP receptor 1A (BMPR1A) in ISCs lead to hyperactivate β-catenin function and loss of BMP signaling, respectively, thus enhancing stem cell features and predisposing mutation carriers to cancer development30,31. TGFβ also promotes differentiation in the apical regions, and inactivating mutations in core TGFβ components are frequently detected in colorectal cancer (CRC). Intriguingly, recent studies propose that the activity of the canonical TGFβ pathway is enhanced by the non-canonical WNT ligand Wnt5A during acute injury in the intestine, and this promotes de novo generation and fission of crypts that facilitate efficient repair of the intestine32,33. At the molecular level, since the Hippo signaling pathway co-transcription factor yes-associated protein (YAP) is also required for crypt regeneration34, interaction between TGFβ and Hippo pathways may explain these observations (see below).
TGFβ signaling in immune regulation
The intestinal epithelium is inhabited by trillions of microbes that are kept in check by diverse epithelial and cellular immune responses35–37. The activity of the immune system, however, has to be tightly controlled to prevent inflammatory disorders and maintain homeostatic regeneration of tissues. TGFβ signaling is involved in the regulation of these processes38–43. Previously, TGFβ signaling was shown to prevent inflammation of the bowel upon activation in the associated dendritic cells44. New data shed further light on this process by suggesting that Clostridium species within the microbiota induce TGFβ1 expression in colonic dendritic cells in a model of experimental colitis45. This in turn stimulates the generation of immunosuppressive regulatory T (Treg) cells, thereby ameliorating the bowel colitis. TGFβ1 also amplifies its own production by activating a Smad3-dependent autocrine signaling loop within dendritic cells, which is, surprisingly, inhibited by Smad2 activity45. Although Smad2 regulates TGFβ receptor expression in this context, it is not clear what drives the opposing roles of the closely related Smad3 and Smad2.
TGFβ signaling was also found to mediate immune-stem cell communication during intestinal repair in Drosophila. Macrophages are recruited to the intestinal epithelium upon injury where they secrete the TGFβ/BMP homologue Decapentaplegic (Dpp)42,46. Dpp then activates the type II receptor Punt (Put) and type I receptor Saxophone (Sax) complex in the ISCs, which drives nuclear localization of dSmad2, thereby triggering ISC proliferation. This immune regulation of ISC function is required for efficient repair of the damaged intestine and is paramount for survival during pathogen invasion. Interestingly, ISCs can subsequently change their response to Dpp by expressing another type I receptor, Thickveins (Tkv), which, upon activation, phosphorylates the R-Smad Mad to restore stem cell quiescence. Oncogenic transformation of ISCs induced by ectopic expression of epidermal growth factor receptor (EGFR) and Jak/STAT pathways can supersede the growth inhibitory effects of Tkv/Mad signaling and requires chronic dSmad2 activity to promote tumor-like stem cell growth and hyperplasia46 (Figure 2). Finally, macrophage-derived Dpp also enhances age-related stem cell dysplasia and epithelial permeability through Sax/dSmad2 signaling46,47. Thus, TGFβ signaling has emerged as an important modulator of immune responses during tissue injury, regeneration, and aging.
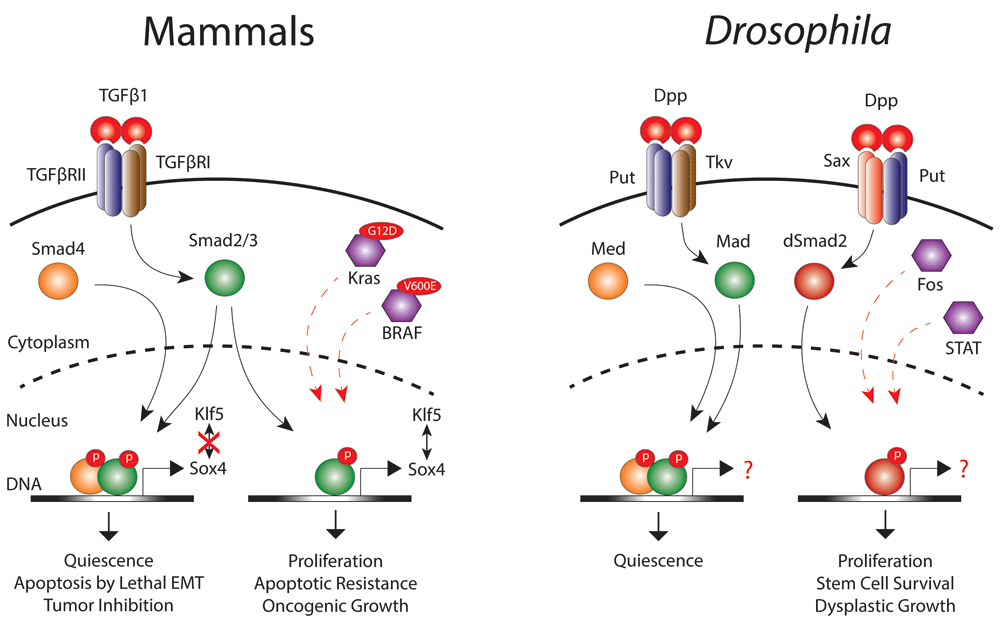
Figure 2. Transforming growth factor β TGFβ signaling during homeostasis and cancer.
In mammals, TGFβ1 activates TGFβ receptors to recruit the canonical Smad2/3–Smad4 complex that, upon nuclear translocation, promotes quiescence and induces lethal epithelial-mesenchymal transition (EMT) to limit tumor growth. Loss of Smad4 in KrasG12D-mutant cells as well as acquisition of a BRAFV600E mutation divert the activity of phosphorylated Smad2/3 to promote cell proliferation, survival, and metastatic growth. Similarly, Drosophila Decapentaplegic (Dpp) stimulates Punt (Put) and Thickveins (Tkv) receptors to activate the canonical Mad-Med (Smad4 homologue) nuclear complex that promotes quiescence. Overactivation of epidermal growth factor receptor (EGFR)/Fos and Jak/signal transducer and activator of transcription (STAT) signaling re-directs Dpp to activate the alternate Put–Saxophone (Sax) receptor complex. This complex then activates dSmad2, which translocates into the nucleus independently of Med and induces tumor-like stem cell growth and dysplasia.
TGFβ signaling in tumor-associated stroma
TGFβ, BMP, and activin signaling play important roles in the crosstalk between tumor cells and the surrounding stroma, although the precise mechanisms are poorly understood48,49. Interestingly, poor prognosis in CRC patients with a stem/serrated/mesenchymal (SSM) subtype was recently linked to the activation of TGFβ signaling within cancer-associated fibroblasts (CAFs), which increased the frequency of tumor-initiating cells and the promotion of metastatic invasion in humanized mouse models50,51. Parallel observations were also made in breast cancer patients where both heat shock factor 1 (HSF1) and TGFβ signaling activation in CAFs is associated with poor clinical outcome52. Another study showed that TGFβ signaling induces a mesenchymal transcriptional footprint in genetically engineered intestinal organoid cultures carrying a BRAFV600E mutation53 (Figure 2). Considering the heterogeneous nature of CRC and the paradoxical roles of TGFβ in stem cell regulation and CRC progression, continued analyses of interactions between mesenchymal and intestinal tissues under normal and dysplastic conditions are required.
A role for Smad4 in switching TGFβ signaling in cancer cells
High TGFβ concentrations are associated with drug resistance and poor prognosis in gastrointestinal cancer patients, yet inactivating mutations in core TGFβ components are frequently found in these maladies. Indeed, deletion of Smad4 in normal intestinal tissues in combination with mutations in APC, TP53, and Kras lead to the formation of invasive carcinoma in mouse xenografts54. Although the immunosuppressive activities of TGFβ1 in the tumor microenvironment have been accepted as one rational explanation, this does not explain the role that TGFβ1 plays within tumor cells lacking functional Smad4 to promote proliferation, metastasis, and apoptotic resistance9,13,14. However, in pancreatic cancer, David et al.55 have now identified Smad4 as a switch between tumor-suppressive and tumor-promoting activities of TGFβ in the tumor cells. Mechanistically, they show that TGFβ induces the expression of Sox4 in a Smad4-independent manner that then cooperates with the epithelial lineage determinant Kruppel like factor 5 (Klf5) to promote oncogenic growth in KrasG12D-driven cancers. Restoration of Smad4 activity disrupts Sox4–KLF5 interaction and redirects Sox4 to induce the transcription of pro-apoptotic genes through a lethal epithelial-mesenchymal transition (EMT) program55 (Figure 2). Thus, Smad4 functions as a transcriptional switch empowering TGFβ1 to control two different transcriptional programs, each leading to opposite outputs. Interestingly, these findings are in agreement with fly studies where dSmad2 was found to promote tumor-like ISC growth and hyperplasia in a Smad4-independent manner46.
The Hippo pathway plays a critical role in cell and organ growth and is implicated in tissue regeneration and cancer34,56–58. In the Hippo pathway, a core kinase cassette comprising MST1/2 and LATS1/2 kinases phosphorylate and thereby sequester the related transcriptional co-activators YAP and transcriptional co-activator with PDZ-binding motif (TAZ) in the cytoplasm, which can also lead to their degradation. Upon inhibition of MST/LATS kinase cassette, YAP/TAZ translocate into the nucleus, where they associate with the DNA-binding partners, such as the TEA domain transcription factors (TEADs), to regulate gene expression. The Hippo pathway is an environmental sensor that responds to a wide range of extrinsic cues that include cell density, cell polarity, metabolic cues, GPCR signaling, and mechanotransduction. The Hippo pathway in turn interacts with several signaling cascades including TGFβ, WNT, EGFR/mitogen-associated protein kinase (MAPK), and others to regulate cell proliferation, tissue growth, apoptotic resistance, and tumor progression56–58. In the case of TGFβ signaling, when the Hippo pathway is inactive, YAP/TAZ physically interact with the Smad2/3–Smad4 complex to promote its nuclear accumulation, thereby regulating diverse transcriptional programs that maintain pluripotency in embryonic stem cells59–61, regulate differentiation into diverse lineages62–67, enhance wound healing68, and promote tumorigenesis in breast cancer, mesothelioma, and hepatocarcinoma models69–71. Recent studies have also uncovered cooperative interactions between YAP and TGFβ-regulated Smads that couple mechanotransduction in fibroblasts to TGFβ-induced profibrotic gene expression programs that are important in fibrosis72,73. Interestingly, the TGFβ pathway can also regulate Hippo signaling by interacting with the scaffold protein RASSF1A, which is often inactivated in human cancers74. Mechanistically, high TGFβ activity promotes ubiquitination-based degradation of RASSF1A. This inactivates the MST/LATS kinase cassette, facilitating nuclear localization of the YAP/Smad complex, thereby enhancing the tumorigenic potential of TGFβ signaling. Together, these studies indicate that the TGFβ and Hippo pathway interactions are finely tuned and disruptions in these processes can alter stem cell function and cell fate decisions and lead to fibrosis and cancer.
TGFβ pathway and hematopoiesis in mammals
TGFβ pathways control several aspects of blood formation21,75. BMPs have been shown to specify hematopoietic tissues during development and promote heterogeneity by differentially regulating the formation of diverse hematopoietic stem cell (HSC) types that are separately deposited in the fetal liver and bone marrow of developing embryos20,21,76. Moreover, BMPs modulate myeloid lineage specification and promote homing and proliferation of hematopoietic progenitors at postnatal stages18,21,77. TGFβ(1–3) ligands, however, are potent inhibitors of hematopoiesis in adults21,78. New studies now identify that megakaryocytes secrete the bulk of latent TGFβ1 in the bone marrow, which is cleaved by the non-myelinating Schwann cells79,80. Once activated, TGFβ1 stimulates canonical Smad2/3 signaling in multipotent HSCs to promote quiescence. Intriguingly, megakaryocyte-derived TGFβ1 also protects HSCs from exhaustion in a hostile microenvironment, which could be relevant to the development of hematological malignancies79. Indeed, leukemia-initiating cells (LICs) in chronic myeloid leukemia (CML) derive therapeutic resistance from active TGFβ signaling81. Although a direct link between megakaryocytes/platelets and the development of hematological malignancies has not been established, platelets are shown to activate TGFβ signaling in circulating tumor cells, enhance immune evasion, and facilitate bone metastasis82,83.
BMP signaling in glioma
Gliomas are the most common tumors of the central nervous system84,85. Among these, diffuse intrinsic pontine gliomas (DIPGs) almost exclusively occur in children at the age of 6–8 years. DIPGs exhibit a dismal prognosis with a median survival rate of only 1 year86 because no therapeutic treatment is effective against these cancers, primarily owing to their anatomical location. Whole genome exploration studies have identified unique epigenetic changes in DIPG specimens with 78% of patients exhibiting a single amino acid mutation in a histone H3 variant, a lysine replacement to methionine at position 27 (K27M). This histone H3-K27M mutation removes trimethylation from the histone H3 tail, allowing transcriptional derepression that is associated with the induction of stem cell features and cancer development87. Intriguingly, four independent studies have now shown that a majority of patients who have the K27M mutation also bear activating mutations in the activin receptor type 1 (ACVR1)88–92. Activity of mutant ACVR1 still requires ligand-mediated activation of type II receptors and does not seem to be tumorigenic alone, since the same ACVR1-activating mutations found in hereditary fibrodysplasia ossificans progressiva (FOP) do not result in cancer predisposition93,94. ACVR1 has not been previously implicated in cancer, thus the link with a mutated histone H3 variant in the context of glioma is particularly intriguing.
Conclusions and perspectives
Extensive efforts in TGFβ research have revealed that dynamic interactions between tissues and rapidly changing molecular contexts determine the specific biological output of TGFβ signaling in intact tissues. These complex processes are designed to fine-tune cellular reactions and prevent exaggerated responses that may lead to lethal consequences in animals. These interactions also allow TGFβ pathways to sense a wide spectrum of constantly changing cellular microenvironments and launch appropriate responses. This complex pattern of tight TGFβ regulation also enables just a few components to control diverse biological activities and creates opportunities for diseased cells to adapt and modify these elements to use for their own growth and survival. Therefore, while it is necessary to keep an open mind when considering such complexities in TGFβ signaling, caution must be exercised when using current tools and interpreting specific observations. This is particularly true in pathological tissues where canonical signaling pathways may be altered. In future work, it will be vital to incorporate information about TGFβ pathway crosstalk and couple observations of pathway activity to mechanistic molecular and functional assays.
All authors contributed to the writing and editing of this article.
Competing interests
The authors declare that they have no competing interests.
Grant information
Work from the authors’ labs is supported by grants from the Canadian Institutes of Health Research (CIHR) Foundation program, the Terry Fox Research Institute, and the Krembil Foundation to LA and JLW. AA is also supported by a postdoctoral fellow award, part of the University of Toronto’s Medicine by Design initiative, which receives funding from the Canada First Research Excellence Fund (CFREF).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgements
We thank past and current members of our laboratories for discussions and contributions towards our understanding of signaling pathways.
Faculty Opinions recommended
References
1.
Huminiecki L, Goldovsky L, Freilich S, et al.:
Emergence, development and diversification of the TGF-beta signalling pathway within the animal kingdom.
BMC Evol Biol.
2009; 9: 28. PubMed Abstract
| Publisher Full Text
| Free Full Text
4.
Mullen AC, Wrana JL:
TGF-β Family Signaling in Embryonic and Somatic Stem-Cell Renewal and Differentiation.
Cold Spring Harb Perspect Biol.
2017; pii: a022186. PubMed Abstract
| Publisher Full Text
5.
Beyer TA, Narimatsu M, Weiss A, et al.:
The TGFβ superfamily in stem cell biology and early mammalian embryonic development.
Biochim Biophys Acta.
2013; 1830(2): 2268–79. PubMed Abstract
| Publisher Full Text
8.
Wakefield LM, Hill CS:
Beyond TGFβ: roles of other TGFβ superfamily members in cancer.
Nat Rev Cancer.
2013; 13(5): 328–41. PubMed Abstract
| Publisher Full Text
9.
Bierie B, Moses HL:
Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer.
Nat Rev Cancer.
2006; 6(7): 506–20. PubMed Abstract
| Publisher Full Text
11.
Lafyatis R:
Transforming growth factor β--at the centre of systemic sclerosis.
Nat Rev Rheumatol.
2014; 10(12): 706–19. PubMed Abstract
| Publisher Full Text
12.
McDonald J, Wooderchak-Donahue W, VanSant Webb C, et al.:
Hereditary hemorrhagic telangiectasia: genetics and molecular diagnostics in a new era.
Front Genet.
2015; 6: 1. PubMed Abstract
| Publisher Full Text
| Free Full Text
16.
Bhatia M, Bonnet D, Wu D, et al.:
Bone morphogenetic proteins regulate the developmental program of human hematopoietic stem cells.
J Exp Med.
1999; 189(7): 1139–48. PubMed Abstract
| Publisher Full Text
| Free Full Text
17.
He XC, Zhang J, Tong WG, et al.:
BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling.
Nat Genet.
2004; 36(10): 1117–21. PubMed Abstract
| Publisher Full Text
18.
Khurana S, Buckley S, Schouteden S, et al.:
A novel role of BMP4 in adult hematopoietic stem and progenitor cell homing via Smad independent regulation of integrin-α4 expression.
Blood.
2013; 121(5): 781–90. PubMed Abstract
| Publisher Full Text
19.
Khurana S, Melacarne A, Yadak R, et al.:
SMAD signaling regulates CXCL12 expression in the bone marrow niche, affecting homing and mobilization of hematopoietic progenitors.
Stem Cells.
2014; 32(11): 3012–22. PubMed Abstract
| Publisher Full Text
| Free Full Text
22.
Weiss A, Attisano L:
The TGFbeta superfamily signaling pathway.
Wiley Interdiscip Rev Dev Biol.
2013; 2(1): 47–63. PubMed Abstract
| Publisher Full Text
23.
Nakao A, Afrakhte M, Morén A, et al.:
Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling.
Nature.
1997; 389(6651): 631–5. PubMed Abstract
| Publisher Full Text
24.
Parker L, Stathakis DG, Arora K:
Regulation of BMP and activin signaling in Drosophila.
Prog Mol Subcell Biol.
2004; 34: 73–101. PubMed Abstract
| Publisher Full Text
25.
Upadhyay A, Moss-Taylor L, Kim MJ, et al.:
TGF-β Family Signaling in Drosophila.
Cold Spring Harb Perspect Biol.
2017; pii: a022152. PubMed Abstract
| Publisher Full Text
26.
Daly AC, Randall RA, Hill CS:
Transforming growth factor beta-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth.
Mol Cell Biol.
2008; 28(22): 6889–902. PubMed Abstract
| Publisher Full Text
| Free Full Text
27.
Grönroos E, Kingston IJ, Ramachandran A, et al.:
Transforming growth factor β inhibits bone morphogenetic protein-induced transcription through novel phosphorylated Smad1/5-Smad3 complexes.
Mol Cell Biol.
2012; 32(14): 2904–16. PubMed Abstract
| Publisher Full Text
| Free Full Text
28.
Beumer J, Clevers H:
Regulation and plasticity of intestinal stem cells during homeostasis and regeneration.
Development.
2016; 143(20): 3639–49. PubMed Abstract
| Publisher Full Text
29.
Crosnier C, Stamataki D, Lewis J:
Organizing cell renewal in the intestine: stem cells, signals and combinatorial control.
Nat Rev Genet.
2006; 7(5): 349–59. PubMed Abstract
| Publisher Full Text
34.
Hong AW, Meng Z, Guan KL:
The Hippo pathway in intestinal regeneration and disease.
Nat Rev Gastroenterol Hepatol.
2016; 13(6): 324–37. PubMed Abstract
| Publisher Full Text
35.
Perez-Lopez A, Behnsen J, Nuccio SP, et al.:
Mucosal immunity to pathogenic intestinal bacteria.
Nat Rev Immunol.
2016; 16(3): 135–48. PubMed Abstract
| Publisher Full Text
41.
Biancheri P, Giuffrida P, Docena GH, et al.:
The role of transforming growth factor (TGF)-β in modulating the immune response and fibrogenesis in the gut.
Cytokine Growth Factor Rev.
2014; 25(1): 45–55. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
42.
Weaver LN, Drummond-Barbosa D:
Gut healing: haemocytes aid via Sax and Tkv jazzes it down.
Nat Cell Biol.
2015; 17(6): 707–9. PubMed Abstract
| Publisher Full Text
48.
Pickup MW, Owens P, Moses HL:
TGF-β, Bone Morphogenetic Protein, and Activin Signaling and the Tumor Microenvironment.
Cold Spring Harb Perspect Biol.
2017; 9(5): pii: a022285. PubMed Abstract
| Publisher Full Text
61.
Beyer TA, Weiss A, Khomchuk Y, et al.:
Switch enhancers interpret TGF-β and Hippo signaling to control cell fate in human embryonic stem cells.
Cell Rep.
2013; 5(6): 1611–24. PubMed Abstract
| Publisher Full Text
65.
Zhang H, von Gise A, Liu Q, et al.:
Yap1 is required for endothelial to mesenchymal transition of the atrioventricular cushion.
J Biol Chem.
2014; 289(27): 18681–92. PubMed Abstract
| Publisher Full Text
| Free Full Text
66.
Narimatsu M, Samavarchi-Tehrani P, Varelas X, et al.:
Distinct polarity cues direct Taz/Yap and TGFβ receptor localization to differentially control TGFβ-induced Smad signaling.
Dev Cell.
2015; 32(5): 652–6. PubMed Abstract
| Publisher Full Text
67.
Yao M, Wang Y, Zhang P, et al.:
BMP2-SMAD signaling represses the proliferation of embryonic neural stem cells through YAP.
J Neurosci.
2014; 34(36): 12039–48. PubMed Abstract
| Publisher Full Text
68.
Lee M, Ran Byun M, Furutani-Seiki M, et al.:
YAP and TAZ regulate skin wound healing.
J Invest Dermatol.
2014; 134(2): 518–25. PubMed Abstract
| Publisher Full Text
69.
Hiemer SE, Szymaniak AD, Varelas X:
The transcriptional regulators TAZ and YAP direct transforming growth factor β-induced tumorigenic phenotypes in breast cancer cells.
J Biol Chem.
2014; 289(19): 13461–74. PubMed Abstract
| Publisher Full Text
| Free Full Text
70.
Fujii M, Toyoda T, Nakanishi H, et al.:
TGF-β synergizes with defects in the Hippo pathway to stimulate human malignant mesothelioma growth.
J Exp Med.
2012; 209(3): 479–94. PubMed Abstract
| Publisher Full Text
| Free Full Text
72.
Szeto SG, Narimatsu M, Lu M, et al.:
YAP/TAZ Are Mechanoregulators of TGF-β-Smad Signaling and Renal Fibrogenesis.
J Am Soc Nephrol.
2016; 27: 3117–28. PubMed Abstract
| Publisher Full Text
| Free Full Text
75.
Cichon MA, Radisky DC:
Cutting the brakes and flooring the gas: how TMEPAI turns TGF-β into a tumor promoter.
Genes Cancer.
2014; 5(9–10): 303–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
84.
Khuong-Quang DA, Buczkowicz P, Rakopoulos P, et al.:
K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas.
Acta Neuropathol.
2012; 124(3): 439–47. PubMed Abstract
| Publisher Full Text
| Free Full Text
87.
Li M, Liu GH, Izpisua Belmonte JC:
Navigating the epigenetic landscape of pluripotent stem cells.
Nat Rev Mol Cell Biol.
2012; 13(8): 524–35. PubMed Abstract
| Publisher Full Text
91.
Zadeh G, Aldape K:
ACVR1 mutations and the genomic landscape of pediatric diffuse glioma.
Nat Genet.
2014; 46(5): 421–2. PubMed Abstract
| Publisher Full Text
92.
Villanueva MT:
Genetics: ACVR1 mutations-a key piece in paediatric diffuse glioma.
Nat Rev Clin Oncol.
2014; 11(6): 300. PubMed Abstract
| Publisher Full Text
93.
Bagarova J, Vonner AJ, Armstrong KA, et al.:
Constitutively active ALK2 receptor mutants require type II receptor cooperation.
Mol Cell Biol.
2013; 33(12): 2413–24. PubMed Abstract
| Publisher Full Text
| Free Full Text
1
Center for Systems Biology, Lunenfeld-Tanenbaum Research Institute, Mount Sinai Hospital, Toronto, ON, Canada 2
Department of Biochemistry and Donnelly Centre, University of Toronto, Toronto, ON, Canada 3
Department of Molecular Genetics, University of Toronto, Toronto, ON, Canada
Work from the authors’ labs is supported by grants from the Canadian Institutes of Health Research (CIHR) Foundation program, the Terry Fox Research Institute, and the Krembil Foundation to LA and JLW. AA is also supported by a postdoctoral fellow award, part of the University of Toronto’s Medicine by Design initiative, which receives funding from the Canada First Research Excellence Fund (CFREF).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Ayyaz A, Attisano L and Wrana JL. Recent advances in understanding contextual TGFβ signaling [version 1; peer review: 2 approved]. F1000Research 2017, 6(F1000 Faculty Rev):749 (https://doi.org/10.12688/f1000research.11295.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles

Comments on this article Comments (0)