Keywords
Plasmodium falciparum, Clinical isolates, Indian Subcontinent, tiling array, array CGH
Plasmodium falciparum, Clinical isolates, Indian Subcontinent, tiling array, array CGH
Plasmodium falciparum is the most virulent among the 6 Plasmodium species that causes malaria in humans. Current efforts to eliminate the disease have not been successful because of the rapid genetic adaptation and natural selection of the parasite. The currently known causes for genome sequence variations in the parasite are single nucleotide polymorphism (SNP), insertions, deletions of short sequences, major amplifications, translocations, inversions, and allelic variations (Cheeseman et al., 2009). Apart from the host factors these parasitic genetic variations may also lead to the disease severity., P.falciparum strains have long been established in continuous culture systems and most genomic analysis has naturally utilized the incredible benefit from such techniques. However, analysis of genome variations from culture adapted parasite lines remains intrinsically very different from analysis of in-vivo derived parasite nucleic acid material and the latter could have a major impact on understanding of the parasite biology (LeRoux et al., 2009). Environmental pressures have been known to introduce changes in genomes providing organisms an evolutionary advantage for adaptation and CNVs could be one such change that assists the parasite adaptation to the new environment. These CNVs could enable survival of the parasite under drug pressure. An amplification event in the GTP cyclohydrolase has been frequently reported and the high copy number of GCH-1 has been shown to confer resistance to anti-folates (Heinberg et al., 2013; Jiang et al., 2008; Kidgell et al., 2006; Nair et al., 2008; Ribacke et al., 2007). The major known parasite molecule that influences the severity of the disease are encoded by Variant Surface Antigen family genes that are located in the sub-telomeric part of the chromosome. P. falciparum 3D7 genome sequencing revealed the presence of ~60 var genes followed by other variant surface antigen families, rifin/stevor (~190), Pfmc-2TM (~12) and surfin (~10) (Frech & Chen, 2013; Scherf et al., 2008). Most studied var genes encodes for PfEMP-1 (Erythrocyte membrane protein-1) that play an important role in the pathogenesis by periodic switching and immune evasion. Along with the exported family members, these genes play an important role in cytoadhesion by binding to multiple cell receptors, including ICAM-1, CD36, E-selectin, NCAM and CD31 on micro vascular endothelia, which mediates sequestration that in-turn, leads to the severity of the disease. Recently another receptor, Endothelial protein C receptor (EPCR), a brain specific receptor has been identified as a binding partner of PfEMP-1 (Smith et al., 2013; Wassmer & Gray, 2017). Var genes are divided into 3 major groups A, B and C and 2 intermediate groups UpsB/A and B/C (Lavstsen et al., 2003). The higher transcript level of var group B and var group C family genes have been reported in severe malaria cases in comparison with the uncomplicated malaria cases from India (Subudhi et al., 2015).
There is limited information regarding complete nuclear genome variation for any of the human malaria parasites from field isolates from the Indian subcontinent, however, Tyagi et al. in 2014 have reported evolutionary history of Plasmodium falciparum by sequence analysis of mitochondrial genome of Indian isolates. Most studies have focused on analysis of sequence variations in single genes from different parts of India (Kumar et al., 2005; Kumar et al., 2012; Mishra et al., 2015; Rajesh et al., 2008; Zeeshan et al., 2012). These have focused on SNP's for "drug resistance" marker genes of Indian isolates. Garg et al., 2009; Pathak et al., 2014; Sharma et al., 2015).
Genomics technologies like whole genome microarrays and more recently next-generation sequencing have enabled deeper analysis of various types of genomic variations. Using a reference clone/isolate, copy number variations of other isolates can be identified (Carret et al., 2005). An interesting study by Oyola et al. in 2016 details of SNP analysis from dried blood spots using Illumina based sequencing approaches.
Array-based technologies have been used to distinguish CNV's in different diseases (Coughlin et al., 2012). Such techniques have been extensively used in cancer research to identify DNA copy number variability to represent structural variations at higher resolution and increased sensitivity (Liu, 2007; Mockler et al., 2005; Przybytkowski et al., 2011).
The objective of this study was to see whether we could at all distinguish differences between the two parasite isolates taken from individual patients showing differing disease severity using aCGH tilling array approach.
The microarray data discussed in this manuscript has been deposited in the NCBI Gene Expression Omnibus (GEO) under the GEO series accession number (GSE77165).
Venous blood samples were collected (5ml) from P. falciparum infected patients admitted to S.P. Medical College, Bikaner, India. Patient’s samples were collected on informed consent according to hospital guidelines. Sample collection was approved by the hospital ethical committee (No.F.(Acad) SPMC/2003/2395). Infection with P. falciparum was confirmed by microscopy and rapid diagnostic tests (OptiMal test; Diamed AG, cressier sur Morat, Switzerland, Falcivax test; Zephyr Biomedical System Goa, India) in the hospital. Blood was immediately (within 15 mins) subjected to density gradient-based separation (Histopaque 1077, Sigma Aldrich, USA) to separate the peripheral blood mononuclear cells (PBMCs) from the infected blood samples following manufactures instruction. Both the fractions were subsequently washed twice with phosphate buffered saline (PBS) and lysed using 4 volumes of TRI-reagent and stored at -80°C. Samples were transported in cold chain to BITS, processed and assessed by 18s r RNA gene based multiplex PCR to eliminate any possibility of P. vivax co-infection. Details of the PCR primers used for multiplex PCR for the detection of P. falciparum and P. vivax are presented in Table 1 as cited in the research articles (Pakalapati et al., 2013). PCR conditions used for the amplifications of 18S r RNA gene involved initial denaturaton at 93°C for 2 minutes followed by denaturation at 93°C for 1 minute 30 seconds, anneal-ing at 52°C for 2 minutes, extension at 72°C for 3 minutes, after initial denaturation all the reactions were run for 30 cycles. The TRI protocol was used to allow investigations of both RNA and DNA from the precious clinical samples. (Chomczynski, 1993) Criteria for determining complicated cases were based on World Health Organization guidelines (World malaria report, 2010). For CGH array hybridization, isolates from two patients were considered a) cerebral malaria (CM) whose Glasgow coma scale = 7/14 was considered as a complicated case (PFC) Pf-2 or test, b) uncomplicated case (PFU) Pf- 1.
| Primer sequence (5'-3') | Detection | Orientation | |
|---|---|---|---|
| 1 | ATC AGC TTT TGA TGT TAG GGT ATT | Genus | Forward |
| 2 | TAA CAA GGA CTT CCA AGC | P. vivax | Reverse |
| 3 | GCT CAA AGA TAC AAA TAT AAG C | P. falciparum | Reverse |
Total DNA was isolated from samples using back extraction buffer following manufacture’s protocol (TRI- Reagent, Sigma Aldrich, and USA). Both the DNA samples (Pf-isolate-2 and Pf-isolate-1) were run on an agarose gel and found to be intact. Total DNA purity and concentration were assessed using Nanodrop ND-1000 UV-Visible spectrophotometer (Nanodrop technologies, Rockland, USA). The A260/A280 ratio suggests the absence of proteins and RNA.
1.5 Micrograms of each DNA sample were restriction digested using Alu1 and Rsa1. This restricted DNA was random-primer labelled with Cyanine-3 (Cy3) and Cyanine-5-dUTP (Cy5) using Agilent Genomic DNA labeling kit (Part Number: 5190-0453). The labelled DNA was concentrated and quality assessed for yields and specific activity. Pf-2 which is a test sample was labelled with Cy3 and Pf-1 sample taken as a control was labelled with Cy5. 5 micrograms of the labelled samples were hybridized on a Genotypic designed P. falciparum 3D7 Custom 2x400K CGH Microarray, (Agilent Microarray Design ID AMADID: 025327). Hybridization of probes was carried out in Agilent Sure hyb chambers at 65°C with rotation for 40 hours. The hybridized slides were washed using Agilent aCGH wash buffer (Part No: 5188-5221/22) and scanned using the Agilent Microarray Scanner G2505C. The Scanned image was processed using Agilent Feature extraction software version 10.7 and probe intensities were normalized using the linear dye normalization method.
A high resolution custom CGH array was designed with 418,577 probes, representing the entire P.falciparum 3D7 reference genome using the e-Array tool SureDesign v 5.0.1 by the Agilent certified microarray facility of Genotypic Technology, Bengaluru, India. Whole genome sequence for P. falciparum 3D7 strain was downloaded from NCBI genome database; the 60mer probes were designed based on optimal GC% and melting temperature using Probe parser V.1.0.exe (Perl program developed by Genotypic Technology, Bangalore, India) Genotypic Probe parser is the perl code developed by Genotypic Technology to design 60mer Probes from the downloaded genome sequence and calculate the GC% and melting temperature (Tm) for the probes. The 60 mer probes designed by the above mentioned program were then subjected to the Agilent eArray tool and the CGH array designed. The probes were designed by tiling over the DNA sequence. The tiling resolution was 56 bp. The probe length has been set as 60 bp which is the optimised length for Agilent comparative genomic hybridization workflow. The probes were designed by calculating GC% and optimum Tm values of (>65 genome sequence and calculate the GC% and melting temperature. The sequence information, including chromosomal location and direction of transcriptional regulation of P. falciparum 3D7 were retrieved from Plasmodium Genome Resource. [PlasmoDB.V.6.0] (Aurrecoechea et al., 2009; Gardner et al., 2002).
Image analysis and data normalization was done by applying the linear dye normalization method using the Agilent Feature Extraction Software version 10.7. The Raw data were analysed using the Agilent Genomic Workbench Software version 6.5. A well-accepted ADM-2 algorithm was used (Aberration Detection Method II algorithm) developed by Agilent and included in the Agilent Genomic Workbench for CGH analysis. ADM-2 is a statistical algorithm which computes copy number differences between the sample and reference using an iterative procedure to identify all genomic regions from its expected value of 0 larger than a given threshold (In this case we have taken a minimum of 5 consecutive probes, with an average log ratio of 0.3). At each iteration, the region with the most significant score is reported. ADM- 2 statistical score is computed as the average normalized Cy3/Cy5 log2 ratios of all probes in the genomic interval multiplied by the square root of the number of these probes. Deletions are reported where the signal intensities of the probes representing the region in the test strain are near background and represents the absence of hybridization. Graphical visualization of the data for the significant aberrations was done using UCSC genome browser (Schneider et al., 2006).
In this study, a custom designed tiling microarray has been used to investigate the overall difference between the two clinical isolates i.e., Pf-isolate-2 (Pf-2) (complicated/cerebral malaria) and Pf-isolate-1 (Pf-1) (uncomplicated malaria). The reference genome 3D7 was used to design the probes. The tiling array used in this study enables large-scale detection of regions showing copy number variations across the whole genome. The CNV's were identified in Pf-2 (complicated) by comparison to Pf-1 (uncomplicated malaria). The well-accepted ADM-2 algorithm was used with filtering criteria of, minimum of 5 probes and the average log fold change of 0.3 for the analysis of CNV. Here we are reporting significant CNV regions seen in Pf-2 in comparison with the Pf-1. In this data, the term amplification and deletion are used to represent copy number of test strain Pf-2 compared to Pf-1. Deletions are reported where the signal intensities of the probes representing the region in the test strain are near background and represent the absence of hybridization.
The size of each chromosome with the number of genes reported here are as per PlasmoDB V.6.0 (Aurrecoechea et al., 2009; Gardner et al., 2002). Those regions that are re-annotated in the genome assembly of P. falciparum 3D7 and updated in recent version of PlasmoDB i.e PlasmoDB V.34 and are eliminated from the study to rule out any inadvertent errors due to the re-annotation processes. A circos plot of whole genome CNV data of Pf-Isolate-2 has been generated to show the regions in chromosomes exhibiting CNV (Figure 1).
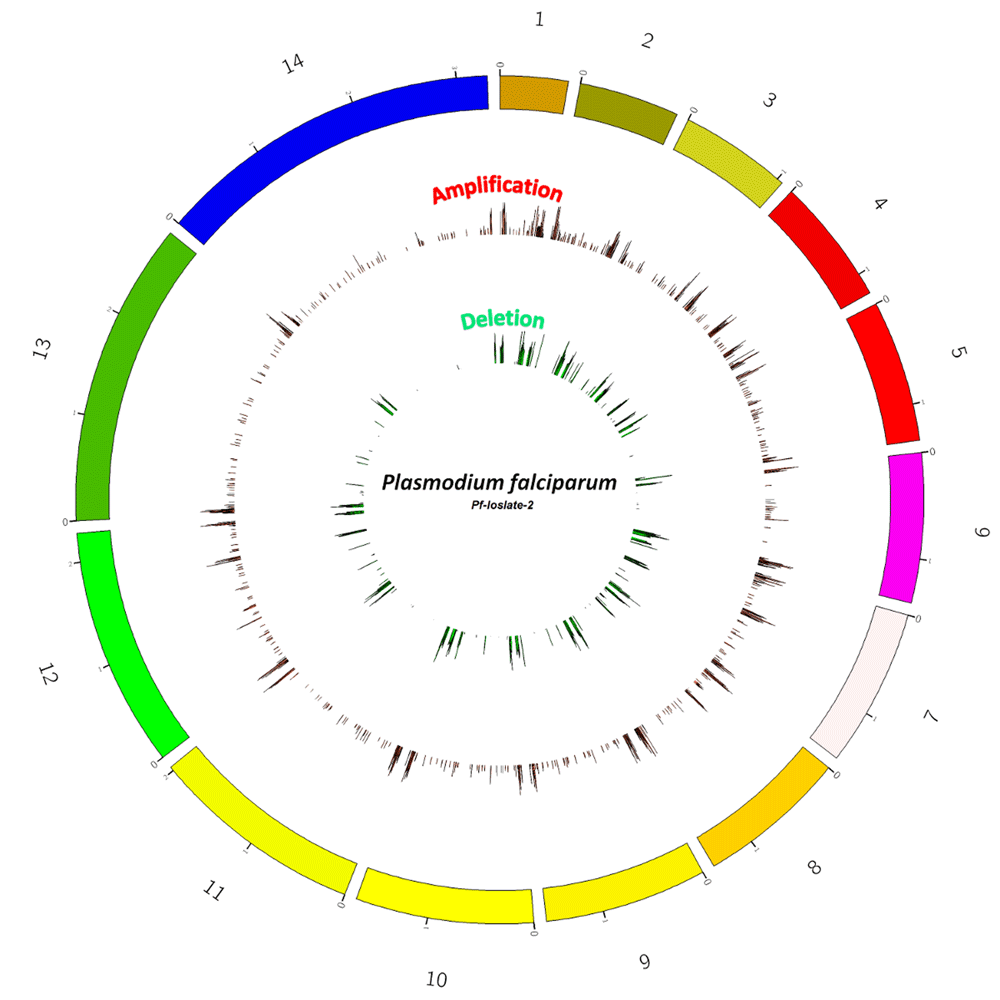
Circle of Genome variation of Plasmodium falciparum (Pf-2), representing probe based chromosome-wise Copy number variations. The outer red bars are showing amplifications i.e. regions with Cy3/ Cy5 ratio of ≥ 0.3 and green inner bars are showing deletions present in the region with Cy3/Cy5 ratio ≤ -0.3.
A total of 687 unique genes was seen to exhibit CNVs in the genome of Pf-isolate-2 (Dataset 1, (Pandey et al., 2018a)). The loss/gains varied in size from 100 nucleotides to 47,000 bp and included intra and intergenic regions, as they are an essential part of the genome and might be involved in the regulation of the genes. From the total of 687 genes, amplification is reported in 511 and deletions in 176 genes. There are 27 genes in regions of chr-4,6,7,8,10,11,12,13 in Pf-2 in which both amplifications/gains and deletions/loss are present. This region is rich in Variant Surface Antigen family genes. The deletions or amplifications covered smaller regions and did not cover more than 2 contiguous genes. Most variations were seen in the sub-telomeric and telomeric regions.
Our analysis revealed CNVs in 268 unique VSA gene families, (Dataset 2, (Pandey et al., 2018b)) out of which variation is present in 58 unique var genes in Pf-2 in comparison to Pf-1. Most of these genes showing CNVs belong to var group B and C. A previous report from our group has also reported differential expression in the transcriptome of these family genes in the isolates causing severe malaria. The longest CNV was reported in chr2: 86, 3297: 94, 7019, near the telomere covering about 20 genes. This region largely had deletions and covered genes belonging to the: var gene family (PfEMP-1) rifin, stevor, and exported family members Plasmodium exported protein (EPF-1, EPF-3, EPF-4) Pfmc-2TM Maurer's cleft two transmembrane protein (MC-2TM) and hypothetical proteins. Regions with multigene family genes showed deletions at both the telomeric and centromeric regions of the chromosomes. In chromosome-1 of Pf-isolate-2 a deletion of 16.3kb (563139-579488) region was seen, that includes 5 genes coding for Stevor, pseudogene (PFA0705C) rifin (RIF, PFA0710C) Plasmodium exported protein (hyp7), unknown function (PFA0715C) Plasmodium exported protein, unknown function, pseudogene (PFA0720W). There is also a 11.7 kb, deletion region in the same chromosome, which includes 4 genes coding for, Exported protein Family-3 (EPF-3, PFA0685C) Exported Protein Family-4 (EPF-4, PFA0690W) Erythrocyte Membrane Protein1 exon2, pseudogene (VAR) (PfEMP-1, VAR, Pseudogene, PFA0695C), and Plasmodium exported protein (hyp10) of unknown function (hyp10, PFA0700C). In previous reports SNPs have been detected in PFA0700C Plasmodium exported protein (hyp10) in malaria parasites from Guinean and Gambian populations (Mobegi et al., 2014). A study by Mok et al. in 2008 have shown a segmental duplication of PFA0675W, PFA0685C, PF0690W and PFA0700C in different strains, and another study from the same group have also reported the duplication of PFA0685C, PF0690W and PFA0695C located at the right end of the chromosome of 3D7 in a clinical isolate (UAM25) (Mok et al., 2008; Ribacke et al., 2007). In our clinical isolate showing complicated manifestation, these segments are showing deletion in the chromosome. A deletion of 11.9 kb region in chromosome-3 that includes conserved Plasmodium proteins with unknown functions (PFC0325C, PFC0330W, PFC0335C). It is interesting to note that although these genes are of unknown function their annotated GO functions suggest that these genes are involved in zinc ion binding activity, receptor activity and in ATP binding activity. A deletion of 14.8 kb, is present in chromosome number 11 and the region includes 6 genes rifin (RIF, PF11_0010), rifin (RIF, PF11_0011), RESA-like protein, pseudogene (RESA pseudogene, PF11_0012) Plasmodium exported protein (PHISTa), unknown function, pseudogene (PHISTa, pseudogene, PF11_0013), Pfmc-2TM Maurer's cleft two transmemberane protein (MC-2TM, PF11_0014) and RESA-like protein, pseudogene, (RESA, PF11_0015). This deletion region is present at the left end of the chromosome. A size variation of 13.2 kb has been seen in chromosome-13 from a starting position of 2828057-2841275 and covers 5 genes rifin (RIF, MAL13P1.495), rifin (RIF, MAL13P1.500), Stevor (Stevor, MAL13P1.505) erythrocyte membrane protein, exon-2, pseudogene (VAR) (PfEMP1, MAL13P1.510) rifin (RIF, MAL13P1.515). Other than larger deletions in chromosomes that cover for VSA, we have seen an amplification of size 13.2 kb present at the right end of chromosome-11 (2006705 - 201923) which covers 5 genes which encodes for rifin (RIF, PF11_0515), Stevor (PF11_0516), rifin (RIF, PF11_0517), rifin (RIF) pseudogene (RIF, PF11_0518) and rifin (RIF, PF11_0519). Details of the mentioned regions, and other regions covering VSA, are shown in Dataset 3 (Pandey et al., 2018c).
On analysis of the CNV data we have detected variations in all the 14 chromosomes; apart from the sub-telomeric regions of the chromosomes, we have also seen variations in other locations on the chromosomes, including intergenic regions.
Other than the regions belonging to multigene family genes, which are mostly showing deletions in the chromosome, there are regions in which amplifications are present and represents genes that encodes for transcriptional co-activator ADA2 (ADA2), chromodomain-helicase-DNA-binding protein 1 homolog, putative (CHD1), polyubiquitin (PfpUB), non-SERCA-type Ca2+ -transporting P-ATPase (ATP4), etc. Major amplifications are present in chromosome-8, 10, and 12. An amplification of 28.6 kb is present in chromosome-8 (249817-278491) (Figure 2), near the centromeric location of the chromosome and covers 3 genes conserved Plasmodium protein, unknown function (MAL8P1139), conserved Plasmodium protein, unknown function (PF08_0122) tRNA pseudouridine synthase, putative (PF08_0123). In chromosome 10, A 7.4 kb and 5.6 kb amplification is present in genes that code for chromodomain helicase DNA binding protein 1 homolog, putative (CHD1, PF10_0232) and transcriptional co-activator ADA-2, (ADA-2, PF10_0143) Another region from 650889-653243 with an amplification of 2.3 kb cover for Glycophorin Binding Protein (GBP, PF10_0159). An amplification of 15.90 kb is present in close proximity to the centromeric region of chromosome 12 (Figure 3), which covers 6 genes that codes for conserved Plasmodium protein with unknown function (PFL1160C), Mitochondrial carrier protein, putative (PFL1145W) conserved plasmodium protein, unknown function (PFL1135C) GTP cyclohydrolase-I GCH1, (GCH-1, PFL1155W) ribosomal protein L24, putative (PFL1150C) and 50S integral memberane protein, putative (PFL1140W) An amplification of the region, that covers three genes, PFL1155W, PFL1150C, and PFL1145W have been seen previously in Dd2 strain of Plasmodium falciparum (Dharia et al., 2009). One of the genes showing amplification codes for the GTP cyclohydrolase enzyme, which is the 1st and the rate-limiting enzyme of the folate biosynthesis pathway. Amplification of this gene has been previously reported in parasite isolates from certain endemic countries (Kidgell et al., 2006; Nair et al., 2008). Another gene, mitochondrial carrier protein, putative (PFL1145W) is reported as a homologue of Yhm2 which was characterized from S. cerevisiae as an inner membrane DNA binding protein. It is speculated that this protein helps in mitochondrial maintenance by tethering mitochondrial DNA to inner membranes (van Dooren et al., 2006). Another region in chromosome-12 showing amplification covers 2 genes codes for Polyubiquitin (PfpUB, PFL0585W) and non-SERCA-type Ca transporting P-ATPase (ATP-4, PFL0590C).
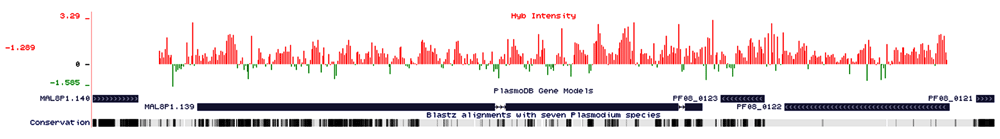
A continuous amplification of 28 kb region (249817-278491) of chromosome-8, the X-axis represents the baseline intensity of the probes, the Y-axis represents the hybridized probe intensity, red bars are showing amplifications present in the region and green bars are showing deletions present in the region. The genes present in the region, are near centromeric location on the chromosome-8 that covers for 3 genes (MAL8P1.139, PF08_0122) conserved Plasmodium membrane protein, unknown function, (PF08_0123) U2 snRNA/tRNA pseudouridine synthase, putative. The white double-headed arrows in single exons are representing the 5the 5exons are representing the 5epresents the the arrows in between the exons are representing introns and strand in which genes are present (Right-hand side arrow forward strand, left-hand side arrow representing reverse strand). The area without any bars are representing no variations.
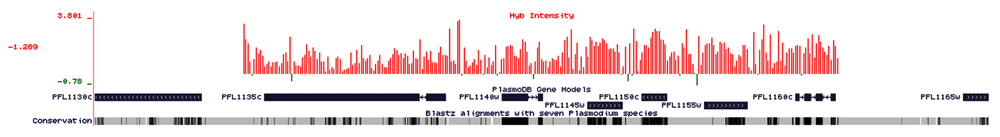
Amplification of 15.90kb is present near close proximity to the centromere region of chromosome 12 (Figure 3), which covers 6 genes, namely (PFL1160c), conserved Plasmodium protein, unknown function, (PFL1145w) mitochondrial carrier protein, putative, (PFL1135c) conserved Plasmodium protein, unknown function, (PFL1155w) GTP cyclohydrolase I (GCH1), (PFL1150c) ribosomal protein L24, putative, (PFL1140w). The X-axis represents the baseline intensity of the probes, the Y-axis represents the hybridized probe intensity, red bars are representing probe intensity as amplifications present in the region and green bars are showing deletions present in the region. Those without any bar are showing no variations.
An amplification of 3.3 kb is present in chromosome -13 (408857- 412219) in the single gene that codes for mRNA-decapping enzyme 2 putative (DCP2, PF13_0048), Amplification of 3.19 present on chromosome-14 covering a single gene coding for pyridine nucelotide transhydrogeanse, putative (PF14_0508) and 3.13 kb which is also present in chromosome-14 covering a single gene EMP-1 trafficking protein (PTP3, PF14_0758). It has been reported that disruption of the gene (PF14_0758) showed reduced levels of expression of PfEMP-1, which suggests that the protein encoded by PF14_0758 plays a role in trafficking and the display of the virulence protein PfEMP-1 on the host erythrocyte (Maier et al., 2008). All these genes showing amplifications in the data are important for the parasite to survive in the host environment.
Several studies at the genomic and transcriptomic level have been undertaken to understand the molecular basis of pathogenesis involved in severe disease manifestation. However, there is scarcity of information on genomic variation in parasites causing complicated manifestations, regions with seasonal malarial transmission. Besides, most of the CNV or SNP studies have been done on in-vitro parasite isolates or clinical isolates from other parts of the world, but very few studies are from parts of India (Cheeseman et al., 2009; Cheeseman et al., 2016). The lacunae in genome variation study still exists as there is no study from parts of India showing nuclear genomic variations between two parasites isolates with differing disease manifestations. CNVs were seen to be present throughout the chromosomes of Pf-2. Large amplifications and deletions of several kilo bases in regions covering Pf-EMP1 and exporter family member genes, have been seen, as it is a known fact that the variant surface antigen family genes are prone to variations and immune invasions. (Carret et al., 2005; Mok et al., 2008; Tan et al., 2009). Amplification in chromosome number 12 includes genes like GCH-1 which is a rate limiting enzyme of folate biosynthesis and is important for the parasite to survive, amplification of this gene is reported to cause antifolate resistance and drives the evolution of drug resistance (Kidgell et al., 2006; Nair et al., 2008). Amplification in Glycophorin binding protein (GBP, PF10_0159) was also seen in the PF-2, this gene is important for the parasite to invade the erythrocyte as it binds to the Glycophorin protein on the erythrocyte membrane. Amplification in sodium-dependent phosphate transporter (PiT, MAL13P1.206) has also seen, the uptake of Pi is essential for the parasite for the synthesis of DNA, RNA and numerous phosphorylated metabolic intermediates (Saliba et al., 2006).
The implications of deletions or amplifications in the coding regions of the genes remain unanswerable at this point. This study shows the presence of CNV based genome differences from two field isolates. Based on msp-1 and msp-2 genotyping it appears that both the isolates exhibit allelic families representing K1 (msp-1 block-2) and FC27 (msp-2 repeats) (Snounou et al., 1999). At this level of the study, it is not possible to speculate whether the differences that we are seeing in Pf-isolate-2 are instrumental in causing the severity of the disease. However, we are reporting here some putative and novel variations, which have not been reported before in single gene studies with field isolates. The Data clearly shows that there is a difference in the isolate which has been taken from the patient with severe malaria (Pf-2) than from that with uncomplicated malaria (Pf-1). Differences exist not only in coding regions, but also in the intergenic locations. Thus, there could be implications at the level of gene regulation. However, these differences could also be inherently natural differences between the two parasite isolates without any direct correlation with the disease states, which could be influenced by various other host factors. This case study is just a preliminary attempt to utilize aCGH tiling array using ADM-2 algorithm to report the variations if any or differences in the genomic segments of parasite isolate causing severe malaria with that of the isolate causing uncomplicated malaria. To get a real picture of the genomic variations related to the disease severity, a tiling array approach can be used to analyze a large set of clinical samples exhibiting different disease complications.
The microarray data discussed in this manuscript has been deposited in the NCBI Gene Expression Omnibus (GEO) under the GEO series accession number (GSE77165).
Dataset 1: Total set of Genes showing Copy Number Variation in Pf-isolate- 2 in comparison with the Pf-isolate- 1. Includes data of total set of genes showing CNV, along with the Information about the Chromosome Number, start of the probe, end of the probe, size of the interval, number of probes in the interval, Amplification call, Deletion call, Gene symbols (PlasmoDB V.6.0), Updated gene symbol (PlasmoDB V.26), Description of gene symbol (PlasmoDB V. 34) log value of the regions, P-value of the region. 10.5256/f1000research.14599.d209655 (Pandey et al., 2018a)
Dataset 2: Copy Number Variations present in Variant Surface Antigen family genes of Pf-isolate-2. Includes data of the total set of VSA gene family genes that are showing variations. Along with the Information about Chromosome Number, start of the probe, end of the probe, the size of the interval, number of probes in the interval, Amplification call, Deletion call, Gene symbols (PlasmoDB V.6.0), Updated gene symbol (PlasmoDB v. 26), Description of gene symbol (PlasmoDB V. 28) log value of the regions, P-value of the region. 10.5256/f1000research.14599.d209656 (Pandey et al., 2018b)
Dataset 3: Genes from Variant Surface Antigen family genes that are showing more than 2 kb variations. Includes data from genes of regions covering for VSA family genes that are showing more than 2 kb variation along with the Information about Chromosome Number, start of the probe, end of the probe, size of the interval, number of probes in the interval, Amplification call, Deletion call, Gene symbols (PlasmoDB V.6.0), Updated gene symbol (PlasmoDB v. 26), Description of gene symbol (PlasmoDB V.34) log value of the regions, p-value of the region. 10.5256/f1000research.14599.d209657 (Pandey et al., 2018c)
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)