Choi SH, Chung SS and Park KS. Re-highlighting the action of PPARγ in treating metabolic diseases [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):1127 (https://doi.org/10.12688/f1000research.14136.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
1Department of Internal Medicine, Seoul National University College of Medicine, Seoul, South Korea 2Department of Internal Medicine, Seoul National University Bundang Hospital, Seongnam, South Korea 3Biomedical Research Institute, Seoul National University Hospital, Seoul, South Korea 4Department of Molecular Medicine and Biopharmaceutical Sciences, Graduate School of Convergence Science and Technology, Seoul National University, Seoul, South Korea
Sung Hee Choi
Roles:
Conceptualization,
Data Curation,
Resources,
Writing – Original Draft Preparation,
Writing – Review & Editing
Sung Soo Chung
Roles:
Conceptualization,
Data Curation,
Resources,
Supervision,
Writing – Original Draft Preparation
Kyong Soo Park
Roles:
Conceptualization,
Data Curation,
Resources,
Supervision,
Writing – Review & Editing
OPEN PEER REVIEW
REVIEWER STATUS
Abstract
Peroxisome proliferator-activated receptor γ (PPARγ) is a member of the nuclear receptor family and plays an important role in adipocyte differentiation, glucose homeostasis, and insulin sensitivity. Thiazolidinediones (TZDs), synthetic ligands of PPARγ, have been used for the treatment of diabetes mellitus for two decades. TZDs were expected to be amazing drugs not only for type 2 diabetes but also for metabolic syndrome and atherosclerotic vascular disease because they can reduce both insulin resistance and inflammation in experimental studies. However, serious unwanted effects pushed TZDs back to an optional second-tier drug for type 2 diabetes. Nevertheless, PPARγ is still one of the most important targets for the treatment of insulin resistance and diabetes mellitus, and novel strategies to modulate PPARγ activity to enhance its beneficial effects and reduce unwanted adverse effects are anticipated. Recent studies showed that post-translational modification (PTM) of PPARγ regulates PPARγ activity or stability and may be a novel way to optimize PPARγ activity with reduced adverse effects. In this review, we will focus on recent advances in PTM of PPARγ and the mechanisms regulating PPARγ function as well as in the development of PPARγ modulators or agonists.
Corresponding author:
Kyong Soo Park
Competing interests:
No competing interests were disclosed.
Grant information:
This work was supported by National Research Foundation Grant by Ministry of Science and ICT, Republic of Korea (NRF-2016R1A2B3010373).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Insulin resistance is the key pathophysiologic abnormality of many metabolic diseases such as type 2 diabetes mellitus, obesity, dyslipidemia, and cardiovascular diseases1. Therefore, reducing insulin resistance is the most important strategy for improving metabolic deterioration. Thiazolidinediones (TZDs), peroxisome proliferator-activated receptor γ (PPARγ) agonists, have shown many beneficial effects not only by enhancing insulin sensitivity but also by demonstrating anti-inflammatory and antioxidant properties, whose actions are related to anti-atherosclerosis2,3. Thus, TZDs were considered a magic bullet for the treatment of type 2 diabetes and atherosclerosis. Indeed, TZDs demonstrated a preventive role for recurrent ischemic stroke in several clinical trials4 and for restenosis after percutaneous coronary intervention (PCI)5–7. However, TZDs increased the risk of peripheral edema, bone loss, and congestive heart failure8–10. A meta-analysis of clinical trials showed that rosiglitazone significantly increased the risk of myocardial infarction11. Although later studies revealed that rosiglitazone did not increase the risk of heart attack and the US Food and Drug Administration (FDA) removed the warning labels from rosiglitazone-containing drugs regarding the issue of increasing heart attack in 2013, rosiglitazone’s cardiovascular safety issue alongside the above-mentioned adverse effects still lead to many physicians hesitating to prescribe TZDs in their clinical practice. Nevertheless, PPARγ is still one of the most important targets for the treatment of insulin resistance and type 2 diabetes, and novel strategies to modulate PPARγ activity to enhance its beneficial effects and reduce unwanted adverse effects are strongly anticipated. Recent studies showed that post-translational modification (PTM) of PPARγ regulates PPARγ activity or stability and may be a novel way to optimize PPARγ activity with reduced adverse effects. In addition, selective PPARγ modulators (sPPARγMs), dual or pan PPAR agonists, have been developed and tested for their metabolic effects in animal studies and in some clinical trials.
PPARγ, a therapeutic target for insulin resistance (Figure 1)
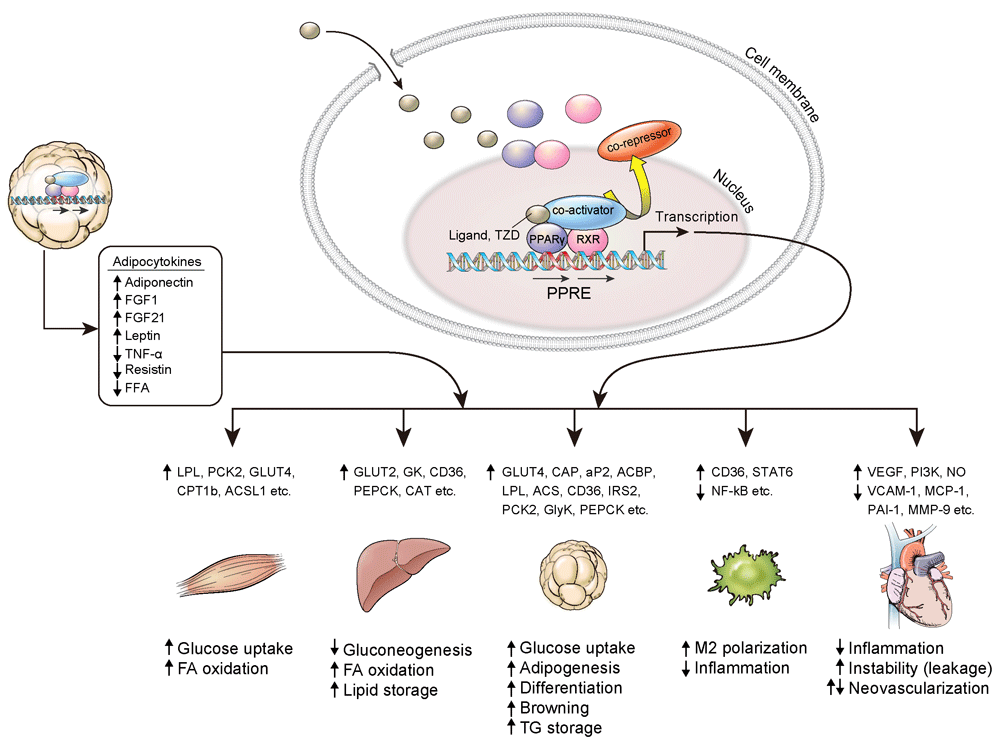
PPARγ is a master regulator of adipocyte differentiation. It is also involved in glucose homeostasis and insulin sensitivity. The expression of PPARγ is most abundant in adipose tissue12. Evidence has shown that the primary target of TZDs is adipose tissue, where it increases the expression of Glut4 and CAP13, and an animal model lacking PPARγ in adipose tissue had a significantly lower response to TZDs14,15. TZDs inhibit the expression of TNF-α, IL-6, and resistin in adipose tissue, which promote insulin resistance and chronic inflammation16,17, while TZDs increased the production of adiponectin and fibroblast growth factor 21 (FGF21), which enhance fatty acid oxidation and insulin sensitivity18,19. TZDs increase lipogenesis by aP2, LPL, CD36, fatty acid transport protein, PEPCK, and the glycerol transporter aquaporin 72 and make adipose tissue store more lipid, while TZDs remove lipid accumulation in other tissues such as muscle and liver20.
Figure 1. Effect of PPARγ activation on various tissues.
ACSL1, acyl-CoA synthetase long chain family member 1; CD36, cluster of differentiation 36; CPT1b, carnitine palmitoyltransferase 1B; FA, fatty acid; FFA, free fatty acid; FGF, fibroblast growth factor; GK, glucokinase; GLUT, glucose transporter; GlyK, glycerol kinase; IRS2, insulin receptor substrate 1; LPL, lipoprotein lipase; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; MCP-1, monocyte chemoattractant protein 1; MMP-9, matrix metalloproteinase 9; NO, nitric oxide; PAI-1, plasminogen activator inhibitor type 1; PCK2, peroxisome proliferator-activated receptor gamma 2 binding site; PEPCK, phosphoenolpyruvate carboxykinase; PI3K, phosphoinositide 3-kinase; PPARγ, peroxisome proliferator-activated receptor γ; PPRE, peroxisome proliferator-activated receptor response element; RXR, retinoid X receptor; STAT6, signal transducer and activator of transcription 6; TG, triglyceride; TNF-α, tumor necrosis factor α; TZD, thiazolidinedione; VCAM-1, vascular cell adhesion molecule-1; VEGF, vascular endothelial growth factor.
From these studies, it seems that improvement of insulin sensitivity in liver and muscle might be secondary to the effects of TZDs in adipose tissue. However, there is also evidence showing that TZDs have an insulin-sensitizing effect on other peripheral organs. It has been demonstrated that ablation of liver PPARγ in mice reduced hepatic steatosis but worsened hyperlipidemia, triglyceride clearance, and muscle insulin resistance21. The expression of PPARγ in skeletal muscle is relatively low compared to adipose tissue, and the physiological significance of PPARγ in skeletal muscle has been shown to work indirectly in previous studies22. However, selective activation of PPARγ in skeletal muscle showed significant protection from high-fat diet-induced insulin resistance and associated changes in muscle phenotype, such as decreasing the quantity of lipid in myocytes and increasing the number of oxidative muscle fiber types23. It suggests that the activation of PPARγ can act directly on muscle tissue to improve insulin sensitivity. Macrophage PPARγ is also implicated in anti-inflammation and lipid metabolism24, and mice lacking macrophage PPARγ are more prone to whole-body insulin resistance25,26.
PPARγ agonists and their effects on the vascular system: friend or foe?
PPARγ is expressed in the endothelium and vascular smooth muscle in the blood vessel wall27,28. Despite controversial cardiovascular effects of TZDs in humans, most experimental studies showed beneficial effects on vascular systems. TZDs inhibit the proliferation and migration of vascular smooth muscle cells (VSMCs), with potential favorable effects on atherosclerosis29,30. Smooth muscle-specific dominant-negative PPARγ transgenic mice showed a loss of nitric oxide responsiveness and high contractility31, which resulted in systolic hypertension. In humans, dominant-negative mutations of PPARγ are associated with early hypertension and insulin resistance32. Activation of PPARγ inhibits CCAAT/enhancer-binding protein-δ (C/EBPδ), which is a well-known mediator of the proinflammatory response in vascular cells33.
TZDs also reduce activation and inflammation in endothelial cells by suppressing the expression of inflammation-associated genes34–37. On the other hand, TZDs induce vascular endothelial growth factor (VEGF) in endothelial cells and increase endothelial cell proliferation and migration by the Akt-dependent pathway38–40. In recent data, rosiglitazone significantly increased endothelial cell migration and vascular leakage in an animal study with increased VEGF expression and suppressed tight junction proteins, which caused instability of the endothelial membrane41. This result could be related to vascular permeability, peripheral edema, and congestive heart failure associated with the use of TZDs, contrary to their beneficial effect on vascular cells. We still need more concrete evidence to understand the role of TZDs in the whole vascular system under various conditions.
Regulation of PPARγ by PTMs to reduce the side effects of TZDs
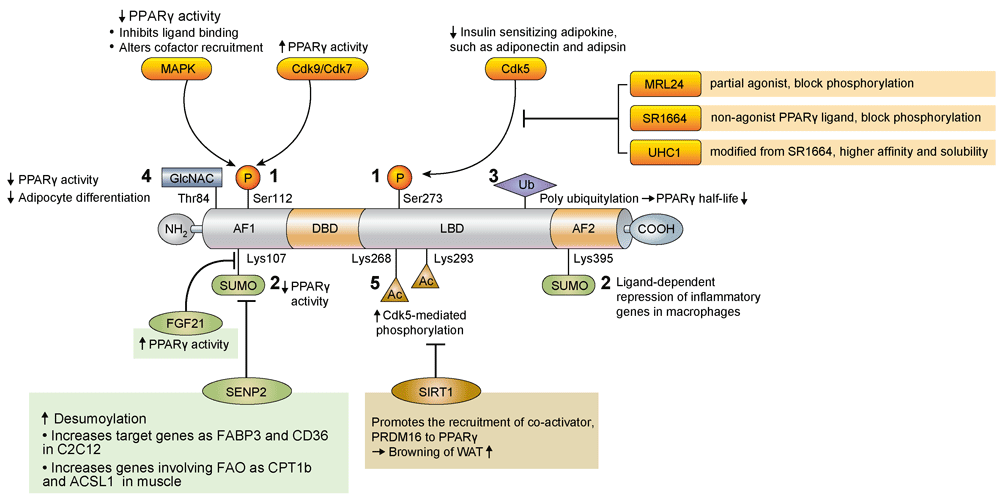
The PTM of PPARγ involves several pathways, including phosphorylation, SUMOylation, ubiquitination, β-O-linked N-acetylglucosamine modification (O-GlcNAcylation), and acetylation. These PTMs are known to regulate both PPARγ expression and its transcriptional activity42 and have been recently suggested as a good modality for reducing the side effects of PPARγ activation by TZDs43 (Figure 2).
Figure 2. Regulation of PPARγ by post translational modification.
Ac, acetyl; ACSL1, acyl-CoA synthetase long chain family member 1; AF1, activation function 1; AF2, activation function 2; CD36, cluster of differentiation 36; Cdk, cyclin-dependent kinase; CPT1b, carnitine palmitoyltransferase 1B; DBD, DNA-binding domain; FABP3, fatty-acid-binding protein 3, muscle and heart; FAO, fatty acid oxidation; FGF, fibroblast growth factor; GlcNAC, N-acetylglucosamine; LBD, ligand-binding domain; Lys, lysine; MAPK, mitogen-activated protein kinase; P, phosphate; PPARγ, peroxisome proliferator-activated receptor γ; PRDM16, PR domain containing 16; SENP2, small ubiquitin-like modifier-specific protease 2; Ser, serine; SIRT1, sirtuin 1; SUMO, small ubiquitin-like modifier; Thr, threonine; Ub, ubiquitin; WAT, white adipose tissue.
Phosphorylation
Phosphorylation at serine 112 (S112) in the N-terminal AF-1 domain was first identified, and various studies revealed that net results of PPARγ phosphorylation may inhibit or stimulate its transcriptional activity depending on the cellular contexts and kinases involved44–48. Phosphorylation at S273 in the ligand-binding domain is mediated by cyclin-dependent kinase 5 (Cdk5), which is activated by pro-inflammatory stimuli and free fatty acids49. S273 phosphorylation affects the expression of insulin-sensitizing adipokines such as adiponectin and adipsin but not those affecting adipogenesis. PPARγ partial agonist MRL24 specifically blocks the phosphorylation of PPARγ at S273 and has higher anti-diabetic activity and fewer side effects than does rosiglitazone49. SR1664 and similar non-agonist PPARγ ligands were also developed for blocking cdk5-mediated phosphorylation and showed improved insulin sensitivity in high-fat diet-fed mice without causing side effects such as fluid retention and weight gain43,50. More recently, it has been reported that phosphorylation at S273 is also facilitated by MEK/ERK, and inhibition of MEK and ERK improves insulin resistance, suggesting that MEK and ERK inhibitors can be therapeutic targets for diabetes through the modulation of PPARγ function51.
SUMOylation
Small ubiquitin-like modifier (SUMO) modification is a reversible process and may affect protein stability, transcriptional activity, and protein–protein interaction. PPARγ is known as a target of SUMOylation. Lysine 107 (K107) of PPARγ2 is the major SUMOylation site, and deSUMOylation of this site increases the transcriptional activity of PPARγ52. The K107R mutant form of PPARγ stimulates adipogenesis and suppresses neointimal formation after balloon injury more effectively than does the PPARγ wild-type form53,54. SUMOylation at K107 of PPARγ may be linked to S112 phosphorylation53. PPARγ SUMOylation at K107 is markedly increased in FGF21-knockout mice, suggesting that FGF21 regulates PPARγ SUMOylation by an unknown mechanism19. SUMOylation of PPARγ at K395 (K365 of PPARγ1) is stimulated by PPARγ agonists, and this modification inhibits the transcription of inflammatory response genes, such as iNOS, through recruiting transcriptional repressors to the NFkB complex in macrophages55.
SUMO-specific protease 2 (SENP2) is the major deSUMOylation enzyme of PPARγ56. Overexpression of SENP2 in C2C12 cells effectively induces PPARγ target genes such as Fabp3 and Cd36 but not Adrp; thus, SENP2 can induce the expression of PPARγ target genes in a selective manner56. SENP2 deSUMOylates PPARγ and PPARδ and activates genes involved in fatty acid oxidation such as Cpt1b and Acsl1, which results in an increase of fatty acid oxidation in muscle. Interestingly, palmitate increases SENP2 expression via the TLR4-MyD88-NFkB pathway. These results suggest that SENP2 is an important regulator of fatty acid metabolism in skeletal muscle57.
Ubiquitination
Ubiquitination is the covalent attachment of ubiquitin, a 76-amino-acid peptide, to lysine residues in the substrate protein. PPARγ has a short half-life and is degraded by the polyubiquitin-proteasome pathway58. Inhibition of proteasome activity by proteasome inhibitors increases PPARγ stability, suggesting ubiquitin modification of PPARγ is an important determinant of PPARγ activity58. Several ubiquitin ligases, such as FBOX9 and Cul4B, and an ubiquitin-specific protease (HAUSP) targeting PPARγ have been identified, and an increase in PPARγ stability generally promotes PPARγ activity and adipogenesis59–61. Interestingly, PPARγ agonists, TZDs, stimulate the ubiquitination of PPARγ, which can be mediated by an ubiquitin ligase, Siah158,62. Therefore, PPARγ ubiquitination may be differently regulated by several ubiquitin E3 ligases or proteases upon various conditions.
O-GlcNAcylation
O-GlcNAcylation is the post-translational cycling of a single β-O-linked N-acetylglucosamine (O-GlcNAc) on the hydroxyl groups of serine or threonine residues of target proteins. A major O-GlcNAc site in PPARγ is T84 in the AF-1 domain in PPARγ2, and increased O-GlcNAcylation reduces its transcriptional activity and adipocyte differentiation63.
Acetylation
Deacetylation at K268 and K293 by the NAD-dependent deacetylase sirtuin 1 (SIRT1) is necessary for the interaction of PPARγ with PRDM16, a transcriptional co-activator for the browning of WAT64. Therefore, SIRT1-dependent PPARγ deacetylation selectively regulates PPARγ activity.
Other PPARγ modulators and agonists
Considering that patients with insulin resistance show many conjugated metabolic problems such as atherosclerosis, obesity, fatty liver, etc., there have been many efforts to develop sPPARγMs, dual or pan PPAR agonists, with potent efficacy but less-deleterious side effects.
sPPARγMs bind to the ligand-binding domain of PPARγ in many ways, which leads to different receptor conformations and cofactor functions65. INT131, a potent non-TZD sPPARγM now in clinical trials, showed excellent glucose lowering with significantly less weight gain, edema with fluid retention, and cardiomegaly than do current TZDs66. Balaglitazone also showed positive effects in the treatment of patients with type 2 diabetes compared to placebo and pioglitazone (phase III clinical study, n = 409), better glycemic control, and less edema compared to pioglitazone 45 mg67. CMHX008 was also tested for its effects in in vitro and in vivo models, showing excellent results by far68.
PPARα/γ dual activation has been the focus of new targets from many pharmaceutical companies, and several clinical trials have been performed in potential treatments such as muraglitazar, tesaglitazar, and aleglitazar. However, owing to unpredictable side effects, the studies were all stopped for further development. Unfortunately, muraglitazar increased cardiovascular events, tesaglitazar increased renal toxicity, and aleglitazar showed bone fractures, heart failure, and gastrointestinal side effects69–71. Recently, (E)-N-(4-(3-(5-bromo-4-hydroxy-2-methoxyphenyl)acryloyl)phenyl)-4-tert-butylbenzamide (SN158) showed anti-diabetic effects through PPARα/γ dual activation. SN158 increased adipogenic differentiation of 3T3-L1 preadipocytes, enhanced fatty acid oxidation in hepatocytes, and increased glucose uptake in myotubes. It lowered plasma glucose and lipid levels in ob/ob mice without severe weight gain. Thus, it represents another candidate PPARα/γ agonist to enhance many metabolic profiles in obesity-related diseases72.
There are some natural products which activate PPARγ and PPARα simultaneously or activate the PPARγ dimer partner retinoid X receptor. Compared to full TZDs, these natural products usually show fewer side effects and comparable anti-diabetic effects73. Honokiol, amorfrutin 1, amorfrutin B, amorphastilbol, genistein, biochanin A, sargaquinoic acid, sargahydroquinoic acid, resveratrol, etc. were tested for their efficacy in in vitro and in vivo studies73.
The development of PPARα/γ/δ pan agonists as anti-diabetic, anti-obesity, or hypolipidemic drugs is still actively ongoing74,75. For example, IVA 337 is a potent and well-balanced pan PPAR agonist which showed promising results in vitro and in vivo and is expected to be used to treat patients with metabolic syndrome and non-alcoholic steatohepatitis76.
Conclusion
PPARγ is still one of the most important targets for the treatment of insulin resistance and diabetes mellitus, even though current use of TZDs in clinical practice is limited because of undesirable adverse effects. Thus, novel strategies to modulate PPARγ activity to enhance its beneficial effects and reduce unwanted side effects have been strongly anticipated. Recent advances in understanding how PTM of PPARγ modulates PPARγ activity provide novel ways to optimize PPARγ activity with reduced adverse effects. In addition, selective PPARγ modulators, dual or pan PPAR agonists, have been developed and tested for their metabolic effects in animal studies and in some clinical trials.
We hope safer PPARγ agonists or modulators with excellent efficacy and fewer adverse effects will be available for treating metabolic diseases and insulin resistance in the near future.
Competing interests
The authors declare that they have no competing interests.
Grant information
This work was supported by National Research Foundation Grant by Ministry of Science and ICT, Republic of Korea (NRF-2016R1A2B3010373).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Faculty Opinions recommended
References
1.
Laakso M, Kuusisto J:
Insulin resistance and hyperglycaemia in cardiovascular disease development.
Nat Rev Endocrinol.
2014; 10(5): 293–302. PubMed Abstract
| Publisher Full Text
2.
Tontonoz P, Spiegelman BM:
Fat and beyond: the diverse biology of PPARgamma.
Annu Rev Biochem.
2008; 77: 289–312. PubMed Abstract
| Publisher Full Text
3.
Ceriello A:
Thiazolidinediones as anti-inflammatory and anti-atherogenic agents.
Diabetes Metab Res Rev.
2008; 24(1): 14–26. PubMed Abstract
| Publisher Full Text
5.
Patel D, Walitt B, Lindsay J, et al.:
Role of pioglitazone in the prevention of restenosis and need for revascularization after bare-metal stent implantation: a meta-analysis.
JACC Cardiovasc Interv.
2011; 4(3): 353–60. PubMed Abstract
| Publisher Full Text
7.
Choi D, Kim SK, Choi SH, et al.:
Preventative effects of rosiglitazone on restenosis after coronary stent implantation in patients with type 2 diabetes.
Diabetes Care.
2004; 27(11): 2654–60. PubMed Abstract
| Publisher Full Text
8.
Berlie HD, Kalus JS, Jaber LA:
Thiazolidinediones and the risk of edema: a meta-analysis.
Diabetes Res Clin Pract.
2007; 76(2): 279–89. PubMed Abstract
| Publisher Full Text
9.
Lago RM, Singh PP, Nesto RW:
Congestive heart failure and cardiovascular death in patients with prediabetes and type 2 diabetes given thiazolidinediones: a meta-analysis of randomised clinical trials.
Lancet.
2007; 370(9593): 1129–36. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
12.
Fajas L, Auboeuf D, Raspé E, et al.:
The organization, promoter analysis, and expression of the human PPARgamma gene.
J Biol Chem.
1997; 272(30): 18779–89. PubMed Abstract
| Publisher Full Text
13.
Ribon V, Johnson JH, Camp HS, et al.:
Thiazolidinediones and insulin resistance: peroxisome proliferatoractivated receptor gamma activation stimulates expression of the CAP gene.
Proc Natl Acad Sci U S A.
1998; 95(25): 14751–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
14.
He W, Barak Y, Hevener A, et al.:
Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle.
Proc Natl Acad Sci U S A.
2003; 100(26): 15712–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
15.
Chao L, Marcus-Samuels B, Mason MM, et al.:
Adipose tissue is required for the antidiabetic, but not for the hypolipidemic, effect of thiazolidinediones.
J Clin Invest.
2000; 106(10): 1221–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
16.
Peraldi P, Xu M, Spiegelman BM:
Thiazolidinediones block tumor necrosis factor-alpha-induced inhibition of insulin signaling.
J Clin Invest.
1997; 100(7): 1863–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
17.
Steppan CM, Bailey ST, Bhat S, et al.:
The hormone resistin links obesity to diabetes.
Nature.
2001; 409(6818): 307–12. PubMed Abstract
| Publisher Full Text
18.
Berg AH, Combs TP, Du X, et al.:
The adipocyte-secreted protein Acrp30 enhances hepatic insulin action.
Nat Med.
2001; 7(8): 947–53. PubMed Abstract
| Publisher Full Text
20.
Ye JM, Dzamko N, Cleasby ME, et al.:
Direct demonstration of lipid sequestration as a mechanism by which rosiglitazone prevents fatty-acid-induced insulin resistance in the rat: comparison with metformin.
Diabetologia.
2004; 47(7): 1306–13. PubMed Abstract
| Publisher Full Text
21.
Gavrilova O, Haluzik M, Matsusue K, et al.:
Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass.
J Biol Chem.
2003; 278(36): 34268–76. PubMed Abstract
| Publisher Full Text
22.
Norris AW, Chen L, Fisher SJ, et al.:
Muscle-specific PPARgamma-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones.
J Clin Invest.
2003; 112(4): 608–18. PubMed Abstract
| Publisher Full Text
| Free Full Text
23.
Amin RH, Mathews ST, Camp HS, et al.:
Selective activation of PPARgamma in skeletal muscle induces endogenous production of adiponectin and protects mice from diet-induced insulin resistance.
Am J Physiol Endocrinol Metab.
2010; 298(1): E28–37. PubMed Abstract
| Publisher Full Text
24.
Wahli W, Michalik L:
PPARs at the crossroads of lipid signaling and inflammation.
Trends Endocrinol Metab.
2012; 23(7): 351–63. PubMed Abstract
| Publisher Full Text
27.
Marx N, Schönbeck U, Lazar MA, et al.:
Peroxisome proliferator-activated receptor gamma activators inhibit gene expression and migration in human vascular smooth muscle cells.
Circ Res.
1998; 83(11): 1097–103. PubMed Abstract
| Publisher Full Text
| Free Full Text
28.
Xin X, Yang S, Kowalski J, et al.:
Peroxisome proliferator-activated receptor gamma ligands are potent inhibitors of angiogenesis in vitro and in vivo.
J Biol Chem.
1999; 274(13): 9116–21. PubMed Abstract
| Publisher Full Text
29.
Li AC, Brown KK, Silvestre MJ, et al.:
Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice.
J Clin Invest.
2000; 106(4): 523–31. PubMed Abstract
| Publisher Full Text
| Free Full Text
30.
Lim S, Lee KS, Lee JE, et al.:
Effect of a new PPAR-gamma agonist, lobeglitazone, on neointimal formation after balloon injury in rats and the development of atherosclerosis.
Atherosclerosis.
2015; 243(1): 107–19. PubMed Abstract
| Publisher Full Text
31.
Halabi CM, Beyer AM, de Lange WJ, et al.:
Interference with PPAR gamma function in smooth muscle causes vascular dysfunction and hypertension.
Cell Metab.
2008; 7(3): 215–26. PubMed Abstract
| Publisher Full Text
| Free Full Text
32.
Barroso I, Gurnell M, Crowley VE, et al.:
Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension.
Nature.
1999; 402(6764): 880–3. PubMed Abstract
| Publisher Full Text
33.
Takata Y, Kitami Y, Yang ZH, et al.:
Vascular inflammation is negatively autoregulated by interaction between CCAAT/enhancer-binding protein-delta and peroxisome proliferator-activated receptor-gamma.
Circ Res.
2002; 91(5): 427–33. PubMed Abstract
| Publisher Full Text
34.
Marx N, Mach F, Sauty A, et al.:
Peroxisome proliferator-activated receptor-gamma activators inhibit IFN-gamma-induced expression of the T cell-active CXC chemokines IP-10, Mig, and I-TAC in human endothelial cells.
J Immunol.
2000; 164(12): 6503–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
35.
Jackson SM, Parhami F, Xi XP, et al.:
Peroxisome proliferator-activated receptor activators target human endothelial cells to inhibit leukocyte-endothelial cell interaction.
Arterioscler Thromb Vasc Biol.
1999; 19(9): 2094–104. PubMed Abstract
| Publisher Full Text
36.
Pasceri V, Wu HD, Willerson JT, et al.:
Modulation of vascular inflammation in vitro and in vivo by peroxisome proliferator-activated receptor-gamma activators.
Circulation.
2000; 101(3): 235–8. PubMed Abstract
| Publisher Full Text
37.
Hong HK, Cho YM, Park KH, et al.:
Peroxisome proliferator-activated receptor gamma mediated inhibition of plasminogen activator inhibitor type 1 production and proliferation of human umbilical vein endothelial cells.
Diabetes Res Clin Pract.
2003; 62(1): 1–8. PubMed Abstract
| Publisher Full Text
38.
Dimmeler S, Dernbach E, Zeiher AM:
Phosphorylation of the endothelial nitric oxide synthase at ser-1177 is required for VEGF-induced endothelial cell migration.
FEBS Lett.
2000; 477(3): 258–62. PubMed Abstract
| Publisher Full Text
39.
Rikitake Y, Kawashima S, Yamashita T, et al.:
Lysophosphatidylcholine inhibits endothelial cell migration and proliferation via inhibition of the extracellular signal-regulated kinase pathway.
Arterioscler Thromb Vasc Biol.
2000; 20(4): 1006–12. PubMed Abstract
| Publisher Full Text
41.
Ku YH, Cho BJ, Kim MJ, et al.:
Rosiglitazone increases endothelial cell migration and vascular permeability through Akt phosphorylation.
BMC Pharmacol Toxicol.
2017; 18(1): 62. PubMed Abstract
| Publisher Full Text
| Free Full Text
42.
van Beekum O, Fleskens V, Kalkhoven E:
Posttranslational modifications of PPAR-gamma: fine-tuning the metabolic master regulator.
Obesity (Silver Spring).
2009; 17(2): 213–9. PubMed Abstract
| Publisher Full Text
43.
Choi SS, Kim ES, Koh M, et al.:
A novel non-agonist peroxisome proliferator-activated receptor γ (PPARγ) ligand UHC1 blocks PPARγ phosphorylation by cyclin-dependent kinase 5 (CDK5) and improves insulin sensitivity.
J Biol Chem.
2014; 289(38): 26618–29. PubMed Abstract
| Publisher Full Text
| Free Full Text
44.
Hu E, Kim JB, Sarraf P, et al.:
Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma.
Science.
1996; 274(5295): 2100–3. PubMed Abstract
45.
Adams M, Reginato MJ, Shao D, et al.:
Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site.
J Biol Chem.
1997; 272(8): 5128–32. PubMed Abstract
| Publisher Full Text
46.
Shao D, Rangwala SM, Bailey ST, et al.:
Interdomain communication regulating ligand binding by PPAR-gamma.
Nature.
1998; 396(6709): 377–80. PubMed Abstract
| Publisher Full Text
47.
Rangwala SM, Rhoades B, Shapiro JS, et al.:
Genetic modulation of PPARgamma phosphorylation regulates insulin sensitivity.
Dev Cell.
2003; 5(4): 657–63. PubMed Abstract
| Publisher Full Text
48.
Compe E, Drané P, Laurent C, et al.:
Dysregulation of the peroxisome proliferator-activated receptor target genes by XPD mutations.
Mol Cell Biol.
2005; 25(14): 6065–76. PubMed Abstract
| Publisher Full Text
| Free Full Text
52.
Ohshima T, Koga H, Shimotohno K:
Transcriptional activity of peroxisome proliferator-activated receptor gamma is modulated by SUMO-1 modification.
J Biol Chem.
2004; 279(28): 29551–7. PubMed Abstract
| Publisher Full Text
53.
Yamashita D, Yamaguchi T, Shimizu M, et al.:
The transactivating function of peroxisome proliferator-activated receptor gamma is negatively regulated by SUMO conjugation in the amino-terminal domain.
Genes Cells.
2004; 9(11): 1017–29. PubMed Abstract
| Publisher Full Text
54.
Lim S, Ahn BY, Chung SS, et al.:
Effect of a peroxisome proliferator-activated receptor gamma sumoylation mutant on neointimal formation after balloon injury in rats.
Atherosclerosis.
2009; 206(2): 411–7. PubMed Abstract
| Publisher Full Text
56.
Chung SS, Ahn BY, Kim M, et al.:
SUMO modification selectively regulates transcriptional activity of peroxisome-proliferator-activated receptor γ in C2C12 myotubes.
Biochem J.
2011; 433(1): 155–61. PubMed Abstract
| Publisher Full Text
57.
Koo YD, Choi JW, Kim M, et al.:
SUMO-Specific Protease 2 (SENP2) Is an Important Regulator of Fatty Acid Metabolism in Skeletal Muscle.
Diabetes.
2015; 64(7): 2420–31. PubMed Abstract
| Publisher Full Text
| Free Full Text
58.
Hauser S, Adelmant G, Sarraf P, et al.:
Degradation of the peroxisome proliferator-activated receptor gamma is linked to ligand-dependent activation.
J Biol Chem.
2000; 275(24): 18527–33. PubMed Abstract
| Publisher Full Text
61.
Lee KW, Cho JG, Kim CM, et al.:
Herpesvirus-associated ubiquitin-specific protease (HAUSP) modulates peroxisome proliferator-activated receptor γ (PPARγ) stability through its deubiquitinating activity.
J Biol Chem.
2013; 288(46): 32886–96. PubMed Abstract
| Publisher Full Text
| Free Full Text
62.
Kilroy G, Kirk-Ballard H, Carter LE, et al.:
The ubiquitin ligase Siah2 regulates PPARγ activity in adipocytes.
Endocrinology.
2012; 153(3): 1206–18. PubMed Abstract
| Publisher Full Text
| Free Full Text
63.
Ji S, Park SY, Roth J, et al.:
O-GlcNAc modification of PPARγ reduces its transcriptional activity.
Biochem Biophys Res Commun.
2012; 417(4): 1158–63. PubMed Abstract
| Publisher Full Text
65.
Higgins LS, Depaoli AM:
Selective peroxisome proliferator-activated receptor gamma (PPARgamma) modulation as a strategy for safer therapeutic PPARgamma activation.
Am J Clin Nutr.
2010; 91(1): 267S–72. PubMed Abstract
| Publisher Full Text
66.
Depaoli AM, Higgins LS, Henry RR, et al.:
Can a selective PPARγ modulator improve glycemic control in patients with type 2 diabetes with fewer side effects compared with pioglitazone?
Diabetes Care.
2014; 37(7): 1918–23. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
67.
Henriksen K, Byrjalsen I, Qvist P, et al.:
Efficacy and safety of the PPARγ partial agonist balaglitazone compared with pioglitazone and placebo: a phase III, randomized, parallel-group study in patients with type 2 diabetes on stable insulin therapy.
Diabetes Metab Res Rev.
2011; 27(4): 392–401. PubMed Abstract
| Publisher Full Text
68.
Ming Y, Hu X, Song Y, et al.:
CMHX008, a novel peroxisome proliferator-activated receptor γ partial agonist, enhances insulin sensitivity in vitro and in vivo.
PLoS One.
2014; 9(7): e102102. PubMed Abstract
| Publisher Full Text
| Free Full Text
69.
Nissen SE, Wolski K, Topol EJ:
Effect of muraglitazar on death and major adverse cardiovascular events in patients with type 2 diabetes mellitus.
JAMA.
2005; 294(20): 2581–6. PubMed Abstract
| Publisher Full Text
70.
Goldstein BJ, Rosenstock J, Anzalone D, et al.:
Effect of tesaglitazar, a dual PPAR alpha/gamma agonist, on glucose and lipid abnormalities in patients with type 2 diabetes: a 12-week dose-ranging trial.
Curr Med Res Opin.
2006; 22(12): 2575–90. PubMed Abstract
| Publisher Full Text
71.
Lincoff AM, Tardif JC, Schwartz GG, et al.:
Effect of aleglitazar on cardiovascular outcomes after acute coronary syndrome in patients with type 2 diabetes mellitus: the AleCardio randomized clinical trial.
JAMA.
2014; 311(5): 1515–25. PubMed Abstract
| Publisher Full Text
73.
Wang L, Waltenberger B, Pferschy-Wenzig EM, et al.:
Natural product agonists of peroxisome proliferator-activated receptor gamma (PPARγ): a review.
Biochem Pharmacol.
2014; 92(1): 73–89. PubMed Abstract
| Publisher Full Text
| Free Full Text
74.
An HJ, Lee B, Kim SM, et al.:
A PPAR Pan Agonist, MHY2013 Alleviates Age-Related Hepatic Lipid Accumulation by Promoting Fatty Acid Oxidation and Suppressing Inflammation.
Biol Pharm Bull.
2018; 41(1): 29–35. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
76.
Boubia B, Poupardin O, Barth M, et al.:
Design, Synthesis, and Evaluation of a Novel Series of Indole Sulfonamide Peroxisome Proliferator Activated Receptor (PPAR) α/γ/δ Triple Activators: Discovery of Lanifibranor, a New Antifibrotic Clinical Candidate.
J Med Chem.
2018; 61(6): 2246–65. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
1
Department of Internal Medicine, Seoul National University College of Medicine, Seoul, South Korea 2
Department of Internal Medicine, Seoul National University Bundang Hospital, Seongnam, South Korea 3
Biomedical Research Institute, Seoul National University Hospital, Seoul, South Korea 4
Department of Molecular Medicine and Biopharmaceutical Sciences, Graduate School of Convergence Science and Technology, Seoul National University, Seoul, South Korea
Sung Hee Choi
Roles:
Conceptualization,
Data Curation,
Resources,
Writing – Original Draft Preparation,
Writing – Review & Editing
Sung Soo Chung
Roles:
Conceptualization,
Data Curation,
Resources,
Supervision,
Writing – Original Draft Preparation
Kyong Soo Park
Roles:
Conceptualization,
Data Curation,
Resources,
Supervision,
Writing – Review & Editing
This work was supported by National Research Foundation Grant by Ministry of Science and ICT, Republic of Korea (NRF-2016R1A2B3010373).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Choi SH, Chung SS and Park KS. Re-highlighting the action of PPARγ in treating metabolic diseases [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):1127 (https://doi.org/10.12688/f1000research.14136.1)
NOTE: If applicable, it is important to ensure the information in square brackets after the title is included in all citations of this article.
track
receive updates on this article
Track an article to receive email alerts on any updates to this article.
Share
Open Peer Review
Current Reviewer Status:
?
Key to Reviewer Statuses
VIEWHIDE
ApprovedThe paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approvedFundamental flaws in the paper seriously undermine the findings and conclusions
I confirm that I have read this submission and believe that I have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
I confirm that I have read this submission and believe that I have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Nagy L and Pap A. Reviewer Report For: Re-highlighting the action of PPARγ in treating metabolic diseases [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):1127 (https://doi.org/10.5256/f1000research.15376.r36080)
NOTE: it is important to ensure the information in square brackets after the title is included in this citation.
Reviewer Report24 Jul 2018
Laszlo Nagy,
SBP Medical Discovery Institute, Florida, USA; Department of Biochemistry and Molecular Biology, University of Debrecen, Debrecen, Hungary
Attila Pap,
Department of Biochemistry and Molecular Biology, University of Debrecen, Debrecen, Hungary
We confirm that we have read this submission and believe that we have an
... Continue reading
Competing Interests: No competing interests were disclosed.
Faculty Reviews are commissioned and written by members of the prestigious Faculty Opinions Faculty, and are edited as a service to our readers. In order to make these reviews as comprehensive and accessible as possible, we seek the reviewers’ input before publication. The reviewers’ names and any additional comments they may have are published alongside the review, as is usual on F1000Research.
We confirm that we have read this submission and believe that we have an appropriate level of expertise to confirm that it is of an acceptable scientific standard.
Nagy L and Pap A. Reviewer Report For: Re-highlighting the action of PPARγ in treating metabolic diseases [version 1; peer review: 2 approved]. F1000Research 2018, 7(F1000 Faculty Rev):1127 (https://doi.org/10.5256/f1000research.15376.r36080)
Alongside their report, reviewers assign a status to the article:
Approved - the paper is scientifically sound in its current form and only minor, if any, improvements are suggested
Approved with reservations -
A number of small changes, sometimes more significant revisions are required to address specific details and improve the papers academic merit.
Not approved - fundamental flaws in the paper seriously undermine the findings and conclusions
Adjust parameters to alter display
View on desktop for interactive features
Includes Interactive Elements
View on desktop for interactive features
Competing Interests Policy
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Examples of 'Non-Financial Competing Interests'
Within the past 4 years, you have held joint grants, published or collaborated with any of the authors of the selected paper.
You have a close personal relationship (e.g. parent, spouse, sibling, or domestic partner) with any of the authors.
You are a close professional associate of any of the authors (e.g. scientific mentor, recent student).
You work at the same institute as any of the authors.
You hope/expect to benefit (e.g. favour or employment) as a result of your submission.
You are an Editor for the journal in which the article is published.
Examples of 'Financial Competing Interests'
You expect to receive, or in the past 4 years have received, any of the following from any commercial organisation that may gain financially from your submission: a salary, fees, funding, reimbursements.
You expect to receive, or in the past 4 years have received, shared grant support or other funding with any of the authors.
You hold, or are currently applying for, any patents or significant stocks/shares relating to the subject matter of the paper you are commenting on.
Stay Updated
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Comments on this article Comments (0)