Introduction
In almost all solid tumours, the single biggest cause of mortality is metastasis1. Metastasis is the spread of tumour cells away from the primary site of origin and subsequent colonisation of distinct secondary sites2. The process of metastasis and the formation of metastases are inherently inefficient3 yet when successful will typically render the cancer incurable1,4,5. Tumour progression to metastasis is not a tumour cell-autonomous program6. It is a multifaceted and complex series of events7, which is influenced at all stages by the intrinsic cellular mutational burden and the numerous bidirectional interactions between malignant and non-malignant cell types and is continuously fine-tuned by the various extrinsic microenvironmental niches, including the biochemistry and biomechanics of the extracellular matrix (ECM)8,9, and availability and activity of growth factors. This process continually evolves depending on the local and distal microenvironments that tumour cells find themselves within or transiting through8,10,11 (Figure 1) and is tuned by inflammation, angiogenesis, lymphangiogenesis, neoneurogenesis12–14, and systemic physiologic stress-responsive pathways such as the sympathetic nervous system15,16. Finally, tumour cells have been known for decades to have the capacity to fuse with one another, leading to further genetic instability, although how this fusion of tumour cells drives the biology of cancer is not yet clear17–19. As a result, our current understanding of how microenvironmental and macroenvironmental cues intersect with intrinsic cancer cell properties to regulate metastatic dissemination is ever-expanding.
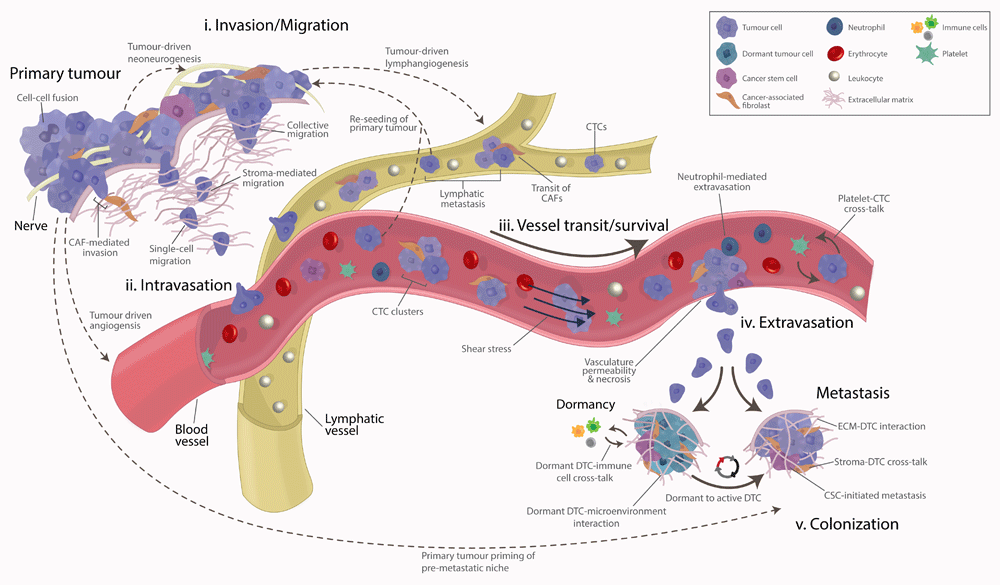
Figure 1. Tumour metastasis is a vast and interconnected array of dynamic and systemic events encompassing both spatial and temporal events.
The process can be broadly divided into the following stages: (i) invasion/migration at/near the primary tumour, (ii) intravasation into the local blood and lymphatic vessels, (iii) survival and transit of cancer cells in the circulation/lymphatics, (iv) arrest and extravasation at secondary sites, and (v) overt colonisation of secondary sites.
Metastasis as a process is one of the most technically challenging aspects of cancer biology to study20–28. As a result, new platforms and technologies are continuously being developed to better understand this process22,29. In this review, we discuss some of the recent advances as well as emerging tools and methodologies being deployed to study metastasis and how the information gleaned is re-shaping our understanding of metastatic dissemination.
The process of metastasis
Metastases, or metastatic disease, is the end result of a vast and interconnected set of dynamic and systemic events encompassing both spatial and temporal selective pressures exerted upon cancer cells30. Over the past few decades, our understanding of these selective pressures and their importance in the various stages of metastatic dissemination has improved significantly. The entire process of metastasis can be broadly divided into the following stages (Figure 1):
(i) invasion/migration at/near the primary tumour
(ii) intravasation into the local blood and lymphatic vessels
(iii) survival and transit of cancer cells in the circulation/lymphatics
(iv) arrest and extravasation at secondary sites
(v) overt colonisation of secondary sites
These different elements (Figure 1) are often seen as distinct yet interconnected progressive stages of a linear cascade, typically associated with the later stages of primary tumour growth. However, we are now beginning to realise that this is far from accurate. Metastatic dissemination can occur from the earliest point of tumourigenesis, prior to the clinical manifestation of tumours31–33, and has been shown to be mediated through processes such as ‘delamination’, whereby cancer cells leave the epithelia and cross the basement membrane34. In many patients, metastasis has already occurred by the time of diagnosis, and, as a result, metastasis prevention may be too late35. Nonetheless, developing a deeper understanding of the process of metastasis which leads to overt metastatic disease, along with the attributes that the cells selected by this process possess, will be critical for treating metastatic disease4 and preventing further metastasis in surgically non-resectable patients. Furthermore, the frequent occurrence of multicellular seeding, whereby multiple primary tumour clones come together to form aggressive polyclonal metastases36 (Figure 2d), and tumour reseeding, whereby circulating tumour cells (CTCs) may return to the primary tumour37, both support the need for continued research into the metastatic process.
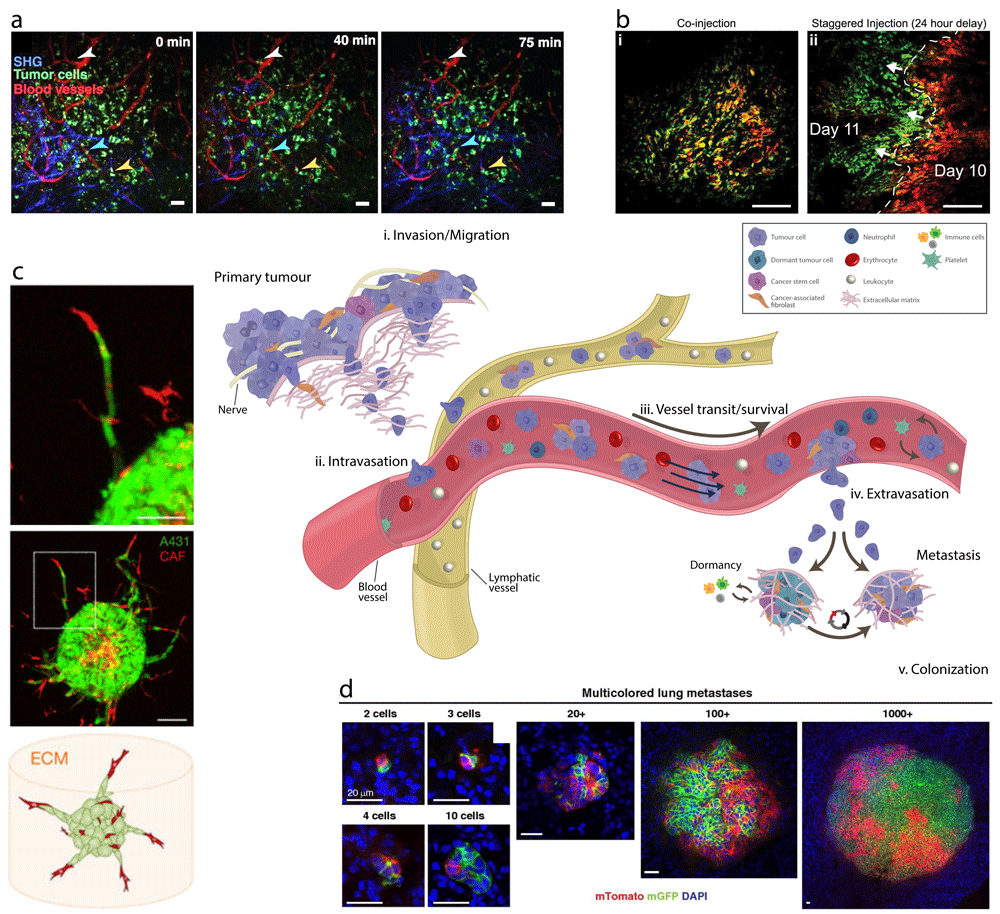
Figure 2. Advances in technologies and tools have allowed us visualise and study some of the stages of metastasis to uncover many of the different mechanisms at play.
(a) Time-lapse intravital imaging of cancer cells (green) in association with blood vessels (red) and collagen fibres—blue, detected by second harmonic generation (SHG)—over the course of 75 minutes shows slow movement (arrowheads) of some cancer cells toward blood vessels. From Pereira et al.69. Reused with permission from the American Association for the Advancement of Science. (b) Tracking the movement of tumour hypoxia using EF5 and pimonidazole probes. Immunofluorescence of KPC xenograft tumours for EF5 (red) and pimonidazole (green) chemical indicators of tumour hypoxia after either (i) co-injection or (ii) 24-hour delayed administration. Images from Conway et al.70, under the terms of the Creative Commons Attribution License (CC BY) (http://creativecommons.org/licenses/by/4.0/). (c) Confocal images of a spheroid (1:1 mixture of cancer-associated fibroblasts [CAFs] [red] and A431 carcinoma [green] cells) after 60 hours of invasion. CAFs (red) lead collective strands of A431 cells (green). Image, originally published in Labernadie et al.71, used with permission from Macmillan Publishers Ltd. (d) Multicellular seeding is a frequent mechanism for distant metastasis. Via Cre recombinase technology, mosaic (red/green) tumour organoids are created and transplanted into non-fluorescent host mice. After 6 to 8 weeks, the lungs of these mice are harvested. Metastases arising exclusively from single-cell seeding produce only single-colour metastases (red OR green). In contrast, multicellular seeding produces metastases with both colours (red AND green). Representative micrographs of polyclonal lung metastases of different sizes from Cheung et al.36. Scale bars = (a) 50 μm and (b, c) 100 μm.
Metastasis involves the selection of traits that are advantageous for the survival of cancer cells. Advances in sequencing platforms38–40 have shown us that micro-evolutionary genetic changes, including somatic mutations, copy number alterations and structural variants in the genome, alongside heritable factors, are detectable independently at both primary and secondary sites as a result of site-specific, context-dependent selective pressures6,33,41–43. This has allowed the identification of hallmark mutational signatures in many different cancer types as well as their metastases44–47 and is facilitating a deeper subclassification of specific cancers48–52. In addition, the precise cancer cell of origin has been shown to heavily influence the trajectory of this entire evolutionary process53. Yet, despite these advances, progress has been painfully slow in translating this genetic information into improved clinical outcomes for patients. As such, more effective translational research to assist in contextualising this genetic information against the concomitant recruitment of traits in the tumour stroma and secondary tissues and organs is required yet is not always easy to achieve54. Such research would allow the dissection of the additional layers of complexity at epigenetic, post-transcriptional, and post-translational levels that regulate expression patterns in different tissue microenvironments. Nonetheless, the concept of metastasis as a successive, linear, and discrete stage-centric process, directed solely by the accumulation of genetic mutations, is flawed and has challenged us to re-examine how we both study and effectively target metastasis and metastases55.
The development of new approaches to detect and quantify sparsely distributed metastatic cells throughout the body at early stages in in vivo tumour models is underway56. However, in the clinical setting, the current tumour staging procedures and even our highest-resolution imaging technologies are not yet sensitive enough to detect micro-metastases or early tumour cell dissemination, the key events in primary tumour progression to metastasis. Similarly, neither in vitro nor in silico tools can accurately recapitulate all stages of metastasis, and more holistic approaches using animal models remain the gold standard21,25,57–59. A new era of translational research is developing, and the insights that it brings are rapidly causing paradigm shifts in our understanding of metastatic phenomena.
Getting things moving: cancer cell migration and invasion
Without question, for metastasis to occur, cancer cells must leave the primary tumour (Figure 1i). This requires the activation and engagement of cellular mechanisms enabling cell movement, adhesion to or degradation of the ECM (or both), and the weakening of cell–cell adhesions to facilitate dissociation from epithelial neighbours. In particular, this centres around actomyosin contractility, which underpins and drives cell migration and invasion60. Cancer invasion is initiated and maintained by signalling pathways (such as the coordinated activity of the RhoGTPases RhoA, Rac1, and Cdc4261) that act to control cytoskeletal dynamics in tumour cells and the turnover of cell–ECM and cell–cell junctions to allow cell migration into the adjacent surrounding tissue (Figure 1i). This process is highly adaptive, being influenced by intrinsic and extrinsic factors, and is typically temporary, having the potential to be reversed. Ultimately, it allows cancer cells to overcome obstacles that would typically impede movement62.
The processes that are activated in cancer cells are similar to those seen in normal cells during embryonic development. These processes allow cancer cells to adapt to their microenvironment and are elicited through changes in cancer cell phenotype and are facilitated, in some situations, by what is known as epithelial-to-mesenchymal transition (EMT)63. The process of EMT is underpinned predominantly by the SNAIL, TWIST, ZEB, and other transcription factor families64,65. In cancer, EMT is thought to play a role in a cancer cell’s acquisition of a stem-like and motile/migratory phenotype, in part through interaction with other important signalling pathways such as the Hippo pathway66. EMT in cancer, however, is not a one-directional permanent program defined by a single pathway63. Instead, it is a partial or reversible process that depends on the intrinsic and extrinsic stimuli that cancer cells receive. This subtle but critical point is what appears to allow cancer cells to undergo both EMT and reciprocal mesenchymal-to-epithelial transition (known as MET) at different stages and locations of the metastatic process67.
The development of new molecular biology approaches and advanced intravital imaging techniques is providing researchers with novel tools for understanding the importance of EMT in cancer progression and metastasis68. There likely exists both EMT-dependent and EMT-independent mechanisms for metastasis, although as yet the specific contexts for each in different cancer types remain elusive. For example, studies on the reversibility of EMT, and in particular the role of EMT markers such as E-cadherin, have shown that fine-tuned modulation of EMT allows switching between stationary and mobile states, whereas others have shown that EMT may be important in cancer stem cell capacity and sensitivity to chemotherapy72–74.
Nonetheless, once acquired, cell movement, broadly speaking, occurs in one of two modes: either individual or collective cell migration75 (Figure 1i). The switch between the two depends heavily on and responds to the physical and molecular triggers present within the microenvironment76. As cancer cells transit within the many different, physiologically distinct, and often hostile multicellular microenvironments, they sense and respond to a plethora of cues, including the biomechanical and biochemical properties of the ECM77,78. In doing so, cancer cells generate both transient and permanent alterations, including ECM remodelling79,80, which leads to the co-evolution of both the cancer cells themselves and the tissues through which they transit81. For example, changes in type I collagen organisation are evident in primary and secondary breast cancer sites82–86, fibronectin levels are altered in ovarian cancer87, and post-translational cross-linking of fibrillar collagens is observed in pancreatic88, breast89,90, and colorectal91,92 cancer, all of which are closely linked to disease progression and metastatic dissemination (Figure 1i).
Interestingly, and perhaps counter-intuitively, the maintenance of epithelial traits during collective cell migration, whereby E-cadherin-dependent cell–cell contacts are maintained, has only recently emerged but has already been shown to be involved in the progression of colorectal cancer93, head and neck squamous cell carcinoma (HNSCC)94, pancreatic cancer95, and breast cancer36,96. An interesting consideration is whether all migrating cancer cells retain such traits or whether indeed fine-tuning of EMT at a population level by the microenvironment is important97 such that only leading cells at the invasive front retain some crucial epithelial traits and acquire new mesenchymal ones98. Whilst detailed investigation into the underlying mechanisms and generation of new tools95,99–102 are underway, it is clear that there is still much we do not know.
Furthermore, in recent years, it has been shown that cancer cell invasion and metastasis are not necessarily cancer cell-autonomous events. Resident stromal cells can be co-opted by cancer cells to facilitate and accelerate processes such as cancer cell invasion. Cancer-associated fibroblasts (CAFs) have been shown to promote cancer cell invasion and metastasis103 through a number of mechanisms, including exerting physical forces on cancer cells via heterotypic E-cadherin/N-cadherin adhesions that enable collective invasion71 (Figure 1i and Figure 2c). In addition, it is known that CAFs heavily influence cancer cell behaviour by inducing processes such as EMT to initiate their invasion104–107 or driving apoptosis to facilitate the switch between expansive invasion and CAF-led invasion108. Thus, the concept that tumours behave as communities109, in which cooperative behaviour occurs not only between cancer cell subclones but also between malignant and non-malignant cells110, adds significantly to the layers of complexity in treating these highly heterogeneous tumours. With this in mind, investigators are undertaking mathematical modelling to better understand the dynamics of cell–cell as well as cell–microenvironmental reciprocities that govern metastatic priming and progression111–114.
A key element that can permit or restrain the invasion of primary tumour cells into the surrounding tissue is the local remodelling of the host microenvironment and in particular the ECM (Figure 1i). Both normal and tumour-associated ECM is deposited, remodelled, and degraded on a continuous basis. However, the tumour-associated ECM in particular is also associated with altered post-translational modification, such as cross-linking, leading to the generation of a dense and usually stiffer fibrotic microenvironment that is pro-tumourigenic11. For years, despite an expanding body of knowledge to the contrary, it was generally believed that this extensive deposition and remodelling of the tumour ECM merely accompanied tumour growth. More recently, however, it has been widely accepted that ECM remodelling is an active contributor to driving cancer progression115 through clustering of integrins and other receptors, leading to downstream activation of intracellular kinase signalling pathways116, which subsequently alter, among other things, EMT and cancer cell migration and invasion117 (Figure 1i).
In addition to changes in the ECM at the primary tumour, there are significant changes in the resident non-malignant cell populations recruited to, or excluded from, the tumour and their activation states. Advances in technologies such as intravital imaging have allowed us to uncover mechanisms by which tumour cells manipulate the normal tissues within which they grow in order to facilitate disease progression59,118,119. Several recent articles have shown the close interplay among CAFs, primary tumour-associated ECM remodelling, and progression of desmoplastic tumours (those surrounded by dense fibrous tissue) such as pancreatic ductal adenocarcinoma88,120–122 and breast cancer123. Most surprising is that CAFs, and the ECM remodelling they underpin, have been shown to play both pro- and anti-tumourigenic roles124,125, highlighting how simple removal of the stroma may not be a suitable therapeutic approach and showing that, instead, subtler approaches such as stromal re-engineering or normalisation, or short-term ‘priming’ interventions120, may represent a more robust approach126,127.
Taking this a step further, there have been several recent studies aimed at mapping the changes in the matrisome (the inventory of all ECM constituents) of primary and metastatic lesions128 in order to generate ECM signatures that could be used to predict outcome and metastasis across different tumours129–131. Because the majority of structural ECM proteins exhibit a remarkable longevity in vivo, often measured in weeks and months and even years8, as opposed to hours for intracellular proteins, we argue that the tumour-specific blend of ECM molecules records a history of tumour evolution132. As such, it has the potential to allow us to better understand how a specific tumour has emerged. Furthermore, this longevity appears to be tissue and tumour type specific. The emergence of ECM signatures to stratify patients with cancer is already providing useful predictors of disease staging130,131, and ECM molecules such as tenascins, periostin, and versicans have been linked to tumour progression and eventually could be used to identify early signs of metastasis133–136. In addition, just as we currently use genomic signatures to identify high-risk patients and predict outcome across a wide array of cancer types, ECM-based proteomic signatures are emerging, and it will not be long before these can be routinely used in the clinic for patient stratification132.
Yet it is not only cells that move around the tumour. Other non-cellular physiological elements such as hypoxia have recently been shown to move within the three-dimensional tumour using dual PLIM/FLIM (phosphorescence lifetime imaging microscopy/fluorescence lifetime imaging microscopy) intravital imaging70 (Figure 2b). The importance of this phenomenon is not to be underestimated, since hypoxia is well known to trigger cell invasion and migration as well as other biological effects such as altered response to therapy70. Thus, daily fluctuations in oxygenation status across the tumour likely reshape the microenvironment, both activating and deactivating signalling pathways and gene expression programs in normal and tumour cells, blunting therapy efficacy and thereby having long-term consequences. Furthermore, it was recently shown that, in breast cancer models, intermittent hypoxia, and not chronic hypoxia, actually promotes clonal diversity and enhances metastatic seeding to secondary organs137.
Going with the flow: intravasation into blood and lymphatics
Intravasation is the active entry of cancer cells into the circulation in order to spread around the body (Figure 1ii and Figure 2a). Logic dictates that it would follow local invasion away from a tumour toward a vessel, and one of the critical requirements for this would be the ability to activate cellular programs that would act to help tumour cells to transverse the endothelial layer of vessels to enter the bloodstream. However, recent evidence has suggested that intratumoural intravasation does not need to be preceded by local invasion and in fact may proceed in parallel to, or independent of, tumour cell invasion into the surrounding stroma138. Either way, intravasation as a process has been incredibly difficult to visualise and model and so has led researchers to believe it is a rare event. This is in contrast to publications showing that, on average, somewhere in the region of 1 million cancer cells per 1 g of tumour tissue can enter and spread daily within the circulation139. Successful studies to date have shown that the escape of cancer cells from the primary tumour into the circulation can occur as both single cells or clusters of a few to a dozen strands or sheets140–143 (Figure 1). What governs the spatial and temporal cues for cancer cell intravasation is still not fully elucidated, but evidence points toward intrinsic cancer cell cues, the activity of stromal cell populations such as macrophages110,144, and organisation of the ECM. For example, cells may orientate according to ECM structures such as collagen fibres that then can direct tumour cell intravasation in in vitro breast cancer cells145.
The vast majority of solid tumours are also able to drive de novo angiogenesis (the growth of new blood vessels) (Figure 1), and malignant progression is typically associated with, and likely even depends on, an angiogenic switch146. Tumour angiogenesis is driven through the secretion of pro-angiogenic growth factors, recruitment of immune cells, and alteration of the perivascular ECM by both tumour cells and associated stromal cells. The generation of leaky tumour vessels is thought to facilitate the dissemination of tumour cells throughout the body and thus represents a viable therapeutic intervention. However, at present, more work is needed to determine whether the majority of intravasation happens predominantly at main vessels or these angiogenic capillary branches. A more comprehensive coverage of angiogenesis in cancer and its therapeutic potential has been reviewed146–148. However, it must be noted that not all solid tumours form or require new vessels for intravasation to occur, and the existence of non-angiogenic tumours is becoming increasingly recognised149.
In addition to metastatic spread through the circulation, an alternative route of dissemination for cancer cells is through the lymphatics (Figure 1ii). Lymphatic metastases may be the preferred route of dissemination for some tumour types such as breast cancer150 and rhabdomyosarcoma151, which, compared with other types of solid tumours, show a higher propensity for lymph node metastasis. Indeed, in many tumour types, the extent of lymph node involvement is a crucial prognostic factor for the disease. Recently, it has been shown that during metastasis, cancer cells escape the primary tumour, intravasate into lymphatic vessels, and reach draining sentinel lymph nodes well before they appear to overtly colonise distant organs via the blood circulation152. This process, shown to occur in mammary carcinoma, squamous cell carcinoma, and melanoma model systems, implicates lymph node metastases as a key step for establishing distant metastases of these tumours69. Nonetheless, metastatic disease as a whole is likely to consist of a complex interplay between disseminating cancer cells exiting the tumours via a combination of both routes, modulated by local and systemic factors and possibly even by sex in cases such as renal clear cell carcinoma where androgen receptor (AR) has been shown to increase haematogenous metastasis yet decrease lymphatic metastasis153.
It has been shown that tumours can also promote lymphangiogenesis (the formation of lymphatic vessels) (Figure 1), which in turn acts to promote cancer cell dissemination154. Aberrant lymphangiogenesis and restructuring of lymphatic networks have been shown to significantly enhance metastasis to both regional lymph nodes and distal organs155 through the secretion of various factors such as vascular endothelial growth factor A/D (VEGF-A/D)156, VEGF-C150,157, interleukin 1 beta (IL-1β)158, fibroblast growth factor (FGF)159, ECM components such as periostin160, or even chronic stress activation of the sympathetic nervous system161.
Whilst lymph node spread of cancer has been known for decades, more recent evidence has implicated the lymphatics not simply as passive highways for tumour cell spread but also as facilitators in many other processes, including the active recruitment of tumour cells to local and distal lymph nodes162 through mechanisms such as CCL21–CCR7 signalling163,164, promoting the survival of metastasising cancer stem cells via CXCL12–CXCR4 signalling165,166, and modulating the host inflammatory response to alter tumour immune surveillance167–170. Inflammation is a critical component of tumour progression, and inflammatory cells are seen as an indispensable participant in progression. Inflammatory cells have been shown to alter cancer cell proliferation, survival, and migration. In some circumstances, cancer cells have co-opted some of the signalling molecules of the innate immune system, including chemokines and their receptors (such as CXCL12–CXCR4 and CCL21–CCR7 mentioned above), to facilitate invasion, migration, and metastasis. As advances in non-invasive imaging technologies improve and allow us to visualise the lymphatics with greater resolution, and with the development of new tools such as the ‘MetAlert’ mice171, which serve to visualise lymphovascular niches in whole animals, we can begin to study the function of tumour-associated lymphatics on metastatic dissemination as well as during therapeutic response.
The road to metastasis: circulating tumour cells
In recent years, an increasing body of evidence has been supporting the primary role for CTCs as the major contributor to metastatic relapse in patients with cancer172 (Figure 1iii). This has fuelled an explosion of interest in their detection and quantification. Indeed, CTCs have been reported in almost all epithelia-derived cancers, including head and neck173, lung174, gastrointestinal (including pancreatic, colorectal and, gastric)175–179, and breast180–187 cancer.
With potentially hundreds of thousands of tumour cells intravasating into the bloodstream, it appears that only a small fraction of CTCs are capable of surviving and extravasating into distant sites to persist as disseminated tumour cells (DTCs)188. Thus, in order for CTCs to become DTCs, they face a number of obstacles that they must overcome to survive whilst transiting within the bloodstream189. Studies have shown that CTCs travel either as individual cells or, more often, as clusters190 (Figure 1iii). These clusters appear in some cases to be heterogeneous in nature, exhibiting combinations of epithelial and mesenchymal traits143. This reintroduces the role of the EMT program in the process of intravasation and cancer cell dissemination. These clusters appear to maintain a partial EMT program which subsequently may facilitate a more robust resistance to apoptosis and an increased propensity to seed and survive at secondary sites191. This resistance to anoikis (apoptosis induced by inadequate or inappropriate cell–cell or cell–ECM interactions) in CTCs has been shown to be driven through various mechanisms, including expression of the tyrosine kinase receptor TrkB192 or activation of non-canonical Wnt signalling193.
Transit within the circulatory system represents one of the most vulnerable times for disseminating cancer cells, and the importance of cooperative host–tumour cell interactions during this time should not be underestimated194 (Figure 1iii). During transit, there is significant cross-talk among tumour cells, accompanying CAFs, platelets, leukocytes, and endothelial cells. These cell–cell contacts and paracrine cell–cell interactions occur both temporally and spatially during transit and at sites of extravasation. For example, CTCs have been shown to associate with activated platelets, which secrete protective signals, such as transforming growth factor-beta (TGF-β), which in turn upregulates nuclear factor kappa B (NFκB) signalling in CTCs, potentially substituting for stromal interactions found at primary and secondary sites195. Furthermore, these platelets have been shown to form protective shields via the deposition of fibrinogen196 and tissue factor (TF)197. In some cases, it has been shown that disseminating tumour cells carry with them primary tumour CAFs along with stromal ECM components198, which subsequently act to facilitate seeding and overt colonisation at secondary sites.
Tools and technologies for the detection of CTCs in the peripheral blood are continuously evolving, yet none has reached the ‘gold’ standard of sensitivity and, more importantly, of specificity172. Nonetheless, every one of these studies supports a critical role for CTCs in metastatic dissemination. To that end, it is now widely accepted that targeting CTCs during haematogenous transport within the circulation may offer an effective approach to targeting the metastatic process, which could lead to the reduction of cancer morbidity and mortality in early stage cancer patients without already-established metastatic diease199.
Of note, one of the major forces that CTCs experience during transit in the circulation is shear stress. The shearing forces exerted on CTCs are caused by the movement of blood over the cell surface. It is heavily influenced by both the viscosity and the velocity of the blood flow200. It is perhaps not surprising that tumour cells have been shown to be more resistant to haemodynamic shear stress than normal cells201,202 and that this feature is crucial not only to survival in the bloodstream but also to the activation of mechanotransduction signalling during attachment at and extravasation into secondary sites203. Furthermore, the generation of tumour microparticles from CTCs attached to vessel walls as a result of shear flow in capillaries within the lung vasculature has been shown to modulate local immune cell behaviour and confer anti-metastatic protection at metastatic sites204.
Next stop, please! Tumour cell extravasation
For decades, it was thought that the specific patterns of metastatic dissemination observed in patients could be explained solely by the dynamics of haematogenous flow7. Not until the 1970s was it demonstrated that regardless of the importance of blood flow, successful metastatic colonisation could occur only at certain organ sites205,206. These studies were the first to provide experimental evidence for organotropic metastasis. Since then, several studies have dissected the various elements of CTC attachment to and extravasation at secondary sites of metastasis. These studies have uncovered critical elements, such as the ability of CTC clusters to manoeuvre through capillary-sized vessels, doing so as a single-cell chain held together through adhesive interactions207. Another study demonstrated that CTC induction of ATP secretion from accompanying activated platelets is able to render the vasculature more permeable by acting on P2Y2 receptors expressed by endothelial cells208. Similarly, CTC-driven platelet-induced alpha-granule secretion contains a wide range of metastasis-promoting growth factors and cytokines that support cancer cell extravasation and survival at secondary sites195.
A recently discovered phenomenon, which has also been shown to play a role in metastasis of some solid tumours, is the production of neutrophil extracellular traps (NETs)209. NETs are extracellular DNA structures that are typically ‘cast’ by neutrophils in response to infection. However, it has been shown that some metastatic cancer cells can stimulate neutrophils to form NETs, which ultimately act to support metastatic colonisation of secondary sites, and these NETs have been observed in both in vivo models and clinical samples210.
Other work has shown that CTC secretion of the CCL2 chemokine is capable of directly inducing vascular permeability211 and subsequently recruiting pro-tumourigenic CCR2 receptor-positive inflammatory monocytes to sites of extravasation212. In addition, the presence of distinct and specific ‘tumour microenvironment of metastasis’ (so-called TMEM in models of breast cancer dissemination) has been described in both genetically engineered models of breast cancer and human breast cancer patients. It has been shown, using intravital microscopy, that a local loss of vascular junctions at TMEMs, mediated by TIE2high macrophage-derived VEGF-A, facilitates cancer cell intravasation and metastasis213 (Figure 1v). Additional factors have been implicated in altering vascular permeability, secreted either locally by tumour cells within the vasculature or systemically from the primary tumour214, to facilitate the alteration of vascular endothelial barriers, including microRNAs (miRs)215, secreted factors such as VEGF, a disintegrin and metalloproteinase domain-containing protein 12 (ADAM12), epiregulin, cyclooxygenase-2, matrix metalloproteinase-1 (MMP-1) and MMP-2216, angiopoietin-like 4217, angiotensin II (ANG-2), MMP-3 and MMP-10218, and finally stromal cell-derived factor 1 (SDF1)219. Many of these factors have also been implicated in the generation of pre-metastatic niches (reviewed elsewhere220). Interestingly, a novel mechanism has recently been described in which CTCs, once arrested on the endothelial wall of blood vessels, can extravasate and coordinate the formation of overt lung metastases via the induction of programmed necrosis (necroptosis) in endothelial cells of vessel walls221. Neutrophils also appear to play an important role in regulating the survival and extravasation of CTCs from the bloodstream, through direct interaction222,223, regulation of natural killer (NK) cell activity via secretion of IL1β or MMPs224 (or both), or altering cytotoxic CD8+ T-cell responses225. Finally, only very recently, researchers have shown that haemodynamic forces and the speed of circulatory flow alone may be critical components of tuning the arrest, adhesion, and extravasation of CTCs from the circulation226 (Figure 1v).
Building new homes: metastatic seeding and tissue colonisation
Despite estimates that over 1 million cancer cells per 1 g of tumour tissue enter the bloodstream daily139, only a very small proportion survive, escape, and become DTCs. An even smaller fraction of these DTCs (that do not become dormant, as discussed in the next section) are capable of progressing toward overt metastases188. It is known that the DTC microenvironment plays an important role in sustaining their survival, regulating their growth, and conferring resistance to therapy227. The ‘seed and soil’ hypothesis proposed by Stephen Paget in 1889 broadly states that colonisation of a secondary site is, in part, dependent on the interactions between tumour cells and the secondary host tissue. That is, inadequate support or cues from secondary tissues, mediated by local resident and recruited cells as well as the ECM, significantly contribute to the inefficiency of the metastatic process.
There is still much discussion as to whether the ability of a tumour cell to overtly colonise a secondary organ is pre-programmed at the primary site prior to leaving or educated upon extravasation at these secondary sites or, more likely, a combination of the two. There is a large body of work addressing how the establishment of pre-metastatic niches and primary tumour-driven remodelling of sites of future metastasis53,228 cross this divide. Work in the PyMT model of breast carcinogenesis has shown that a rare population of primary tumour-derived cancer stem cells can initiate metastases in the lung and that, accordingly, the ability of these tumours to metastasise is dependent on the induction of periostin expression in secondary sites in order to maintain cancer cell stemness229. Furthermore, the oxygen-rich environment in the lung may act to restrain T-cell responses to extravasating cancer cells and induce tolerance to provide a more hospitable environment for metastatic colonisation230. Similarly, the ability of DTCs to physically interact with the ECM, at least in the context of the lung, appears to be contingent upon their ability to form filopodium-like protrusions that are rich in integrin beta-1231. DTCs that are unable to sense or respond to these secondary organ cues thus fail to activate the proliferative programs, driven primarily by FAK, SRC, and ERK signalling, that are necessary for overt metastatic colonisation92,232. As such, it has been shown that targeting Src and ERK signalling pathways may be a potential therapeutic approach to block overt metastatic colonisation of the lung by breast cancer cells233.
Overt colonisation of tissues likely requires a series of tissue-specific events, which may explain the propensity of certain tumours for metastatic organotropism. For example, in the brain, DTCs encounter reactive astrocytes that produce plasminogen activator, which leads to the production of plasmin and induces DTC death. The ability of DTCs to survive in this hostile environment is therefore dependent upon the ability of the cancer cell to express serpins234, which typically are produced by neurons and protect against plasminogen activator-mediated cell death235. Conversely, serpins have also been shown to be important in stromal remodelling and local invasion at the primary tumour in pancreatic cancer236, highlighting tissue- and context-dependent roles for this family across multiple stages of metastatic dissemination. Thus, the ability of DTCs to acquire or express markers of non-malignant resident cells in tissues, and in doing so mimicking these cells, could be a malignant adaptation required for survival and overt secondary organ colonisation. In another example, a study has shown that metastasising breast cancer cells arriving in the brain display a GABAergic phenotype similar to that of neuronal cells, which enhances their survival and subsequent metastatic colonisation237.
In addition to cell-intrinsic properties of the arriving cancer cells, their ability to subvert resident stromal cells to initiate remodelling programs in these new and distinct environments, such as the bone238, is critical in facilitating overt colonisation. Thus, the local stroma, comprising ECM, non-malignant cells, and the signalling molecules they produce, is an integral and vital component of secondary niches that, together with the underlying genetic aberrations in the cancer cells, determines the growth characteristics, morphology, and aggressiveness of disseminating tumour cells239. For example, lung colonisation by breast cancer cells is enhanced by the deposition of the ECM components tenascin C133 and periostin229 or post-translational cross-linking of collagens86.
It is now well established that sites of future metastasis within secondary organs are not merely the passive receivers of CTCs but instead are selectively and actively modified by the primary tumour prior to the arrival of CTCs220. The term pre-metastatic niche was coined over a decade ago to describe the systemic modification of secondary tissue microenvironments to facilitate subsequent metastatic colonisation by disseminating tumour cells240. In order to maximise the chance of overt metastatic colonisation of secondary organs, the combined action of tumour-secreted factors and tumour-shed extracellular vesicles (cargo-containing vesicles that are secreted by cells into the extracellular space and can bind to and be incorporated into other target cells to facilitate cell–cell communication) is required to facilitate this pre-metastatic niche formation. Together, their coordinated action induces changes such as the induction of vascular leakiness241, remodelling of stroma and ECM90,242, along with systemic effects on the immune system. Many of these secreted factors are transported within cancer exosomes (extracellular vesicles, typically 40 to 100 nm, shed from the surface of cells) possessing unique surface marker compositions, which act to facilitate guiding of the exosomes and their cargo to specific secondary organs of future metastasis243–246. For example, pancreatic cancer cell-secreted exosomes have been shown to accumulate in secondary tissues such as the liver and lead to the generation of pre-metastatic niches through activating hepatic stellate cells and Kupffer cells to drive ECM remodelling243 and can be detected in the circulating blood, offering promise of potential biomarker applications. Given the technical limitations of studying these early pre-metastatic events in vivo and in the clinic, there has recently been a push to develop engineered niche-mimicking biomaterials to better study this process247,248.
Lying low: disseminated tumour cell dormancy
Dormancy is defined as the latent state in which (tumour) cells remain quiescent and are reversibly arrested in the G0 phase of the cell cycle188. When tumour cells enter a patient’s bloodstream, the cells transit to and lodge in various microenvironments such as niches in the lung tissue or bone marrow. Upon arrival, the tumour cells may become dormant. These dormant tumour cells can spend months, years, and even decades in these niches, which act as a safe haven, in many cases providing protection from adjuvant therapies28. Dormant tumour cells are typically seen as chemotherapy-resistant because they are not actively dividing; however, the molecular mechanisms underlying this resistance are still poorly understood227. There are also emerging arguments in the field that, rather than wait for these metastases to emerge before initiating treatments, it may be more effective to target the dormant metastatic seeds or their dormancy-inducing niches before they re-awaken (or both)227 or, perhaps more controversially, actively stimulate their re-awakening during adjuvant therapy.
In some situations, studies of metastatic tumour dissemination have shown that primary tumour-driven mechanisms act to counter the overt colonisation of secondary tissues and thereby induce dormancy. For example, rather than forming pre-metastatic niches (discussed above) that act to increase the efficiency of metastatic colonisation, tumours may create specialised microenvironments in which tumour cells can become quiescent, allowing DTCs to survive in a dormant state. These ‘sleepy niches’ or ‘silent’ pre-metastatic niches220 result in the extensive delay in the development of overt metastasis. DTCs thus appear to be able to persist long term within organs, re-awakening many months or years later when the host organs inevitably succumb to overt colonisation249. Of note, in addition, there is experimental evidence to show that DTCs can persist in other organs that rarely develop metastases250. However, what governs the re-awakening of dormant DTCs is still the topic of much debate.
An important factor determining the persistence of dormant DTCs appears to be their ability to escape the body’s immune surveillance. Previous work has shown that DTCs can evade NK cell clearance by decreasing the expression of NK ligands, a program that appears to be tightly coupled with their entry into a quiescent state251. Similarly, it appears that, in some cases, DTCs can be held in a state of dormancy by CD4+ and CD8+ T cells252–254. There are also several factors that have been shown to induce or sustain (or both) the dormancy of DTCs in secondary tissues and they tend to be organ specific. For example, bone morphogenetic protein 4 (BMP4) is present in many tissues, yet elevated levels in the lung contribute to modulating prostate255 and breast cancer256 cell dormancy. Breast cancer cells lodged within the bone marrow can activate Src signalling and expression of the CXCR4 receptor which in turn activates pro-survival signalling in response to bone-derived CXCL12257. Similarly, in the bone marrow, secreted factors such as BMP7 and TGF-β2, as well as ECM components such as secreted protein acidic and rich in cysteine (SPARC), have been shown to modulate HNSCC258 and prostate cancer259,260 cell dormancy. Thrombospondin-1 (TSP1) produced from mature endothelial cells and deposited into the microvascular basement membrane is able to confine DTCs to a quiescent state in some tissues261. However, given the ubiquitous nature of TSP1 in other tissues, it strongly indicates that a co-operative interaction with other factors present within each tissue-specific context may be at play. In this particular study, the authors elegantly dissect the role of vascular niches, demonstrating that TSP1 suppresses DTC outgrowth in both the lung and the bone marrow but not in the brain261.
Both inflammation and ECM remodelling programs elicit profound effects on cell behaviour, including DTCs and cellular dormancy programs. The outgrowth of previously dormant DTCs in the lung has been shown to be activated by both inflammation262 and TGF-β-driven fibrotic type I collagen remodelling263. Similarly, other tissue-resident cells, including osteoblasts and osteoclasts, have been shown to control the switching of dormancy programs within the endosteal niche in multiple myeloma264.
Clinical translation and implications
Clinically, metastatic disease represents a major challenge and is responsible for more than 90% of deaths associated with solid tumours265. Conventional drugs for cancer treatment are largely cytostatic drugs aimed at targeting intrinsic cancer cell mechanisms such as cell cycle progression. Although in many instances they are successful in reducing the size of primary tumours, they have been shown to have little effect on DTCs, potentially owing to the increased heterogeneity and significant mutational burden of DTCs, which facilitates efficient evasion of cell death1,265,266. Furthermore, evidence suggests that some chemotherapies may trigger metastasis through increasing intravasation267. Thus, much research has turned to finding drugs which interfere with cell motility, targeting phases such as cancer cell invasion and migration through the surrounding ECM265, intravasation, and extravasation.
Treating cancer metastasis is further challenged by the logistical and indeed ethical difficulties in evaluating metastasis formation and development in clinical trials. Running metastasis-preventing trials on patients with early stage cancer using survival and reduction of metastases as the endpoint is not always viable, as these studies will be lengthy and will require a large number of patients with otherwise relatively good survival prospects. One must also remember that the metastatic pathway is a dynamic, ongoing process, which has, in many patients, already occurred before primary diagnosis, meaning that successful treatment would require targeting of early or already-established metastasis rather than the initial process of dissemination and colonisation. Similarly, it is highly unlikely that a single metastasis-preventing agent will be maximally effective, and so co-targeting multiple elements of the metastatic process, coupled with new clinical trial designs, is required, though not always readily achievable, to improve patient outcome and improve survival. However, given the current landscape, there is still much work needed before successful targeting of established metastasis can become a clinical reality.
Grant information
The authors are supported by the National Health and Medical Research Council of Australia, Susan G. Komen for the Cure (CCR17483294), National Breast Cancer Foundation, Cancer Institute NSW, the Australian Research Council, a Len Ainsworth Pancreatic Cancer Fellowship, Cancer Council NSW, St Vincent’s Clinic Foundation, Sydney Catalyst, and Tour de Cure. This project was made possible by an Avner Pancreatic Cancer Foundation grant.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgements
We apologise to the authors whose work could not be discussed here. We would like to thank Christine Chaffer for critical reading and advice.
Faculty Opinions recommendedReferences
- 1.
Steeg PS:
Tumor metastasis: mechanistic insights and clinical challenges.
Nat Med.
2006; 12(8): 895–904. PubMed Abstract
| Publisher Full Text
- 2.
Steeg PS:
Targeting metastasis.
Nat Rev Cancer.
2016; 16(4): 201–18. PubMed Abstract
| Publisher Full Text
- 3.
Chambers AF, Groom AC, MacDonald IC:
Dissemination and growth of cancer cells in metastatic sites.
Nat Rev Cancer.
2002; 2(8): 563–72. PubMed Abstract
| Publisher Full Text
- 4.
Gupta GP, Massagué J:
Cancer metastasis: building a framework.
Cell.
2006; 127(4): 679–95. PubMed Abstract
| Publisher Full Text
- 5.
Eccles SA, Welch DR:
Metastasis: recent discoveries and novel treatment strategies.
Lancet.
2007; 369(9574): 1742–57. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 6.
Valastyan S, Weinberg RA:
Tumor metastasis: molecular insights and evolving paradigms.
Cell.
2011; 147(2): 275–92. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 7.
Lambert AW, Pattabiraman DR, Weinberg RA:
Emerging Biological Principles of Metastasis.
Cell.
2017; 168(4): 670–91. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 8.
Cox TR, Erler JT:
Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer.
Dis Model Mech.
2011; 4(2): 165–78. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 9.
Yue B:
Biology of the extracellular matrix: An overview.
J Glaucoma.
2014; 23(8 Suppl 1): S20–3. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 10.
Wong GS, Rustgi AK:
Matricellular proteins: priming the tumour microenvironment for cancer development and metastasis.
Br J Cancer.
2013; 108(4): 755–61. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 11.
Cox TR, Erler JT:
Molecular pathways: connecting fibrosis and solid tumor metastasis.
Clin Cancer Res.
2014; 20(14): 3637–43. PubMed Abstract
| Publisher Full Text
- 12.
Lolas G, Bianchi A, Syrigos KN:
Tumour-induced neoneurogenesis and perineural tumour growth: a mathematical approach.
Sci Rep.
2016; 6: 20684. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 13.
Ayala GE, Dai H, Powell M, et al.:
Cancer-related axonogenesis and neurogenesis in prostate cancer.
Clin Cancer Res.
2008; 14(23): 7593–603. PubMed Abstract
| Publisher Full Text
- 14.
Magnon C, Hall SJ, Lin J, et al.:
Autonomic nerve development contributes to prostate cancer progression.
Science.
2013; 341(6142): 1236361. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 15.
Kim TH, Rowat AC, Sloan EK:
Neural regulation of cancer: from mechanobiology to inflammation.
Clin Transl Immunology.
2016; 5(5): e78. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 16.
Pimentel MA, Chai MG, Le CP, et al.:
Sympathetic Nervous System Regulation of Metastasis. Madame Curie Bioscience Database - NCBI Bookshelf. 2013. Reference Source
- 17.
Bastida-Ruiz D, van Hoesen K, Cohen M:
The Dark Side of Cell Fusion.
Int J Mol Sci.
2016; 17(5): pii: E638. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 18.
Lu X, Kang Y:
Cell fusion as a hidden force in tumor progression.
Cancer Res.
2009; 69(22): 8536–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 19.
Searles SC, Santosa EK, Bui JD:
Cell-cell fusion as a mechanism of DNA exchange in cancer.
Oncotarget.
2018; 9(5): 6156–73. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 20.
Sahai E:
Illuminating the metastatic process.
Nat Rev Cancer.
2007; 7(10): 737–49. PubMed Abstract
| Publisher Full Text
- 21.
Gómez-Cuadrado L, Tracey N, Ma R, et al.:
Mouse models of metastasis: progress and prospects.
Dis Model Mech.
2017; 10(9): 1061–74. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 22.
Sleeboom JJF, Eslami Amirabadi H, Nair P, et al.:
Metastasis in context: modeling the tumor microenvironment with cancer-on-a-chip approaches.
Dis Model Mech.
2018; 11(3): pii: dmm033100. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 23.
Wright LE, Ottewell PD, Rucci N, et al.:
Murine models of breast cancer bone metastasis.
Bonekey Rep.
2016; 5: 804. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 24.
Dai J, Hensel J, Wang N, et al.:
Mouse models for studying prostate cancer bone metastasis.
Bonekey Rep.
2016; 5: 777. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 25.
Francia G, Cruz-Munoz W, Man S, et al.:
Mouse models of advanced spontaneous metastasis for experimental therapeutics.
Nat Rev Cancer.
2011; 11(2): 135–41. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 26.
Fantozzi A, Christofori G:
Mouse models of breast cancer metastasis.
Breast Cancer Res.
2006; 8(4): 212. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 27.
Saxena M, Christofori G:
Rebuilding cancer metastasis in the mouse.
Mol Oncol.
2013; 7(2): 283–96. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 28.
Croucher PI, McDonald MM, Martin TJ:
Bone metastasis: the importance of the neighbourhood.
Nat Rev Cancer.
2016; 16(6): 373–86. PubMed Abstract
| Publisher Full Text
- 29.
Malandrino A, Kamm RD, Moeendarbary E:
In Vitro Modeling of Mechanics in Cancer Metastasis.
ACS Biomater Sci Eng.
2018; 4(2): 294–301. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 30.
Cox TR, Gartland A, Erler JT:
The pre-metastatic niche: is metastasis random?
Bonekey Rep.
2012; 1: 80. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 31.
Harper KL, Sosa MS, Entenberg D, et al.:
Mechanism of early dissemination and metastasis in Her2+ mammary cancer.
Nature.
2016; 540: 588–562. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 32.
Hosseini H, Obradović MM, Hoffmann M, et al.:
Early dissemination seeds metastasis in breast cancer.
Nature.
2016; 540: 552–558. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 33.
Klein CA:
Parallel progression of primary tumours and metastases.
Nat Rev Cancer.
2009; 9(4): 302–12. PubMed Abstract
| Publisher Full Text
- 34.
Rhim AD, Mirek ET, Aiello NM, et al.:
EMT and dissemination precede pancreatic tumor formation.
Cell.
2012; 148(1–2): 349–61. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 35.
Talmadge JE, Fidler IJ:
AACR centennial series: the biology of cancer metastasis: historical perspective.
Cancer Res.
2010; 70(14): 5649–69. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 36.
Cheung KJ, Padmanaban V, Silvestri V, et al.:
Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters.
Proc Natl Acad Sci U S A.
2016; 113(7): E854–63. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 37.
Kim MY, Oskarsson T, Acharyya S, et al.:
Tumor self-seeding by circulating cancer cells.
Cell.
2009; 139(7): 1315–26. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 38.
Lavin Y, Kobayashi S, Leader A, et al.:
Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses.
Cell.
2017; 169(4): 750–765.e17. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 39.
Nakagawa H, Wardell CP, Furuta M, et al.:
Cancer whole-genome sequencing: Present and future.
Oncogene.
2015; 34(49): 5943–50. PubMed Abstract
| Publisher Full Text
- 40.
Petljak M, Alexandrov LB:
Understanding mutagenesis through delineation of mutational signatures in human cancer.
Carcinogenesis.
2016; 37(6): 531–40. PubMed Abstract
| Publisher Full Text
- 41.
de Velasco G, Wankowicz SA, Madison R, et al.:
Targeted genomic landscape of metastases compared to primary tumours in clear cell metastatic renal cell carcinoma.
Br J Cancer.
2018; 118(9): 1238–42. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 42.
Gerlinger M, Rowan AJ, Horswell S, et al.:
Intratumor heterogeneity and branched evolution revealed by multiregion sequencing.
N Engl J Med.
2012; 366(10): 883–92. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 43.
Gerlinger M, Horswell S, Larkin J, et al.:
Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing.
Nat Genet.
2014; 46(3): 225–33. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 44.
Alexandrov LB, Nik-Zainal S, Wedge DC, et al.:
Signatures of mutational processes in human cancer.
Nature.
2013; 500(7463): 415–21. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 45.
Morganella S, Alexandrov LB, Glodzik D, et al.:
The topography of mutational processes in breast cancer genomes.
Nat Commun.
2016; 7: 11383. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 46.
Bailey P, Chang DK, Nones K, et al.:
Genomic analyses identify molecular subtypes of pancreatic cancer.
Nature.
2016; 531(7592): 47–52. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 47.
Chitale D, Gong Y, Taylor BS, et al.:
An integrated genomic analysis of lung cancer reveals loss of DUSP4 in EGFR-mutant tumors.
Oncogene.
2009; 28(31): 2773–83. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 48.
Cancer Genome Atlas Research Network, Linehan WM, Spellman PT, et al.:
Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma.
N Engl J Med.
2016; 374(2): 135–45. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 49.
Ricketts CJ, De Cubas AA, Fan H, et al.:
The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma.
Cell Rep.
2018; 23(1): 313–326.e5. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 50.
Cancer Genome Atlas Research Network: The Molecular Taxonomy of Primary Prostate Cancer.
Cell.
2015; 163(4): 1011–25. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 51.
Cancer Genome Atlas Network: Comprehensive molecular portraits of human breast tumours.
Nature.
2012; 490(7418): 61–70. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 52.
Waddell N, Pajic M, Patch AM, et al.:
Whole genomes redefine the mutational landscape of pancreatic cancer.
Nature.
2015; 518(7540): 495–501. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 53.
Visvader JE:
Cells of origin in cancer.
Nature.
2011; 469(7330): 314–22. PubMed Abstract
| Publisher Full Text
- 54.
Sleeman J, Steeg PS:
Cancer metastasis as a therapeutic target.
Eur J Cancer.
2010; 46(7): 1177–80. PubMed Abstract
| Publisher Full Text
- 55.
Steeg PS, Theodorescu D:
Metastasis: a therapeutic target for cancer.
Nat Clin Pract Oncol.
2008; 5(4): 206–19. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 56.
Kubota SI, Takahashi K, Nishida J, et al.:
Whole-Body Profiling of Cancer Metastasis with Single-Cell Resolution.
Cell Rep.
2017; 20(1): 236–50. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 57.
Chen S, Sanjana NE, Zheng K, et al.:
Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis.
Cell.
2015; 160(6): 1246–60. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 58.
Gengenbacher N, Singhal M, Augustin HG:
Preclinical mouse solid tumour models: status quo, challenges and perspectives.
Nat Rev Cancer.
2017; 17(12): 751–65. PubMed Abstract
| Publisher Full Text
- 59.
Nobis M, Warren SC, Lucas MC, et al.:
Molecular mobility and activity in an intravital imaging setting - implications for cancer progression and targeting.
J Cell Sci.
2018; 131(5): pii: jcs206995. PubMed Abstract
| Publisher Full Text
- 60.
Pandya P, Orgaz JL, Sanz-Moreno V:
Actomyosin contractility and collective migration: may the force be with you.
Curr Opin Cell Biol.
2017; 48: 87–96. PubMed Abstract
| Publisher Full Text
- 61.
Timpson P, McGhee EJ, Morton JP, et al.:
Spatial regulation of RhoA activity during pancreatic cancer cell invasion driven by mutant p53.
Cancer Res.
2011; 71(3): 747–57. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 62.
Pandya P, Orgaz JL, Sanz-Moreno V:
Modes of invasion during tumour dissemination.
Mol Oncol.
2017; 11(1): 5–27. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 63.
Brabletz T, Kalluri R, Nieto MA, et al.:
EMT in cancer.
Nat Rev Cancer.
2018; 18(2): 128–34. PubMed Abstract
| Publisher Full Text
- 64.
Kalluri R, Weinberg RA:
The basics of epithelial-mesenchymal transition.
J Clin Invest.
2009; 119(6): 1420–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 65.
Heerboth S, Housman G, Leary M, et al.:
EMT and tumor metastasis.
Clin Transl Med.
2015; 4: 6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 66.
Lehmann W, Mossmann D, Kleemann J, et al.:
ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types.
Nat Commun.
2016; 7: 10498. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 67.
Brabletz T:
To differentiate or not--routes towards metastasis.
Nat Rev Cancer.
2012; 12(6): 425–36. PubMed Abstract
| Publisher Full Text
- 68.
Zhao Z, Zhu X, Cui K, et al.:
In Vivo Visualization and Characterization of Epithelial-Mesenchymal Transition in Breast Tumors.
Cancer Res.
2016; 76(8): 2094–104. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 69.
Pereira ER, Kedrin D, Seano G, et al.:
Lymph node metastases can invade local blood vessels, exit the node, and colonize distant organs in mice.
Science.
2018; 359(6382): 1403–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 70.
Conway JRW, Warren SC, Herrmann D, et al.:
Intravital Imaging to Monitor Therapeutic Response in Moving Hypoxic Regions Resistant to PI3K Pathway Targeting in Pancreatic Cancer.
Cell Rep.
2018; 23(11): 3312–26. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 71.
Labernadie A, Kato T, Brugués A, et al.:
A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion.
Nat Cell Biol.
2017; 19(3): 224–37. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 72.
Beerling E, Seinstra D, de Wit E, et al.:
Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity.
Cell Rep.
2016; 14(10): 2281–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 73.
Zheng X, Carstens JL, Kim J, et al.:
Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer.
Nature.
2015; 527(7579): 525–30. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 74.
Fischer KR, Durrans A, Lee S, et al.:
Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance.
Nature.
2015; 527(7579): 472–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 75.
Friedl P, Locker J, Sahai E, et al.:
Classifying collective cancer cell invasion.
Nat Cell Biol.
2012; 14(8): 777–83. PubMed Abstract
| Publisher Full Text
- 76.
van Helvert S, Storm C, Friedl P:
Mechanoreciprocity in cell migration.
Nat Cell Biol.
2018; 20(1): 8–20. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 77.
Lu P, Weaver VM, Werb Z:
The extracellular matrix: a dynamic niche in cancer progression.
J Cell Biol.
2012; 196(4): 395–406. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 78.
Pickup MW, Mouw JK, Weaver VM:
The extracellular matrix modulates the hallmarks of cancer.
EMBO Rep.
2014; 15(12): 1243–53. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 79.
He X, Lee B, Jiang Y:
Cell-ECM Interactions in Tumor Invasion.
Adv Exp Med Biol.
2016; 936: 73–91. PubMed Abstract
| Publisher Full Text
- 80.
Kai F, Laklai H, Weaver VM:
Force Matters: Biomechanical Regulation of Cell Invasion and Migration in Disease.
Trends Cell Biol.
2016; 26(7): 486–97. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 81.
Friedl P, Alexander S:
Cancer invasion and the microenvironment: plasticity and reciprocity.
Cell.
2011; 147(5): 992–1009. PubMed Abstract
| Publisher Full Text
- 82.
Provenzano PP, Eliceiri KW, Campbell JM, et al.:
Collagen reorganization at the tumor-stromal interface facilitates local invasion.
BMC Med.
2006; 4(1): 38. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 83.
Huo CW, Chew G, Hill P, et al.:
High mammographic density is associated with an increase in stromal collagen and immune cells within the mammary epithelium.
Breast Cancer Res.
2015; 17: 79. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 84.
McConnell JC, O'Connell OV, Brennan K, et al.:
Increased peri-ductal collagen micro-organization may contribute to raised mammographic density.
Breast Cancer Res.
2016; 18(1): 5. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 85.
Maller O, Hansen KC, Lyons TR, et al.:
Collagen architecture in pregnancy-induced protection from breast cancer.
J Cell Sci.
2013; 126(Pt 18): 4108–10. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 86.
Cox TR, Bird D, Baker AM, et al.:
LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis.
Cancer Res.
2013; 73(6): 1721–32. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 87.
Kenny HA, Chiang CY, White EA, et al.:
Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion.
J Clin Invest.
2014; 124(10): 4614–28. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 88.
Miller BW, Morton JP, Pinese M, et al.:
Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy.
EMBO Mol Med.
2015; 7(8): 1063–76. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 89.
Erler JT, Bennewith KL, Nicolau M, et al.:
Lysyl oxidase is essential for hypoxia-induced metastasis.
Nature.
2006; 440(7088): 1222–6. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 90.
Erler JT, Bennewith KL, Cox TR, et al.:
Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche.
Cancer Cell.
2009; 15(1): 35–44. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 91.
Baker AM, Bird D, Lang G, et al.:
Lysyl oxidase enzymatic function increases stiffness to drive colorectal cancer progression through FAK.
Oncogene.
2013; 32(14): 1863–8. PubMed Abstract
| Publisher Full Text
- 92.
Baker AM, Cox TR, Bird D, et al.:
The role of lysyl oxidase in SRC-dependent proliferation and metastasis of colorectal cancer.
J Natl Cancer Inst.
2011; 103(5): 407–24. PubMed Abstract
| Publisher Full Text
- 93.
Chung YC, Wei WC, Hung CN, et al.:
Rab11 collaborates E-cadherin to promote collective cell migration and indicates a poor prognosis in colorectal carcinoma.
Eur J Clin Invest.
2016; 46(12): 1002–11. PubMed Abstract
| Publisher Full Text
- 94.
Veracini L, Grall D, Schaub S, et al.:
Elevated Src family kinase activity stabilizes E-cadherin-based junctions and collective movement of head and neck squamous cell carcinomas.
Oncotarget.
2015; 6(10): 7570–83. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 95.
Erami Z, Herrmann D, Warren SC, et al.:
Intravital FRAP Imaging using an E-cadherin-GFP Mouse Reveals Disease- and Drug-Dependent Dynamic Regulation of Cell-Cell Junctions in Live Tissue.
Cell Rep.
2016; 14(1): 152–67. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 96.
Ye X, Tam WL, Shibue T, et al.:
Distinct EMT programs control normal mammary stem cells and tumour-initiating cells.
Nature.
2015; 525(7568): 256–60. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 97.
Pietilä M, Ivaska J, Mani SA:
Whom to blame for metastasis, the epithelial-mesenchymal transition or the tumor microenvironment?
Cancer Lett.
2016; 380(1): 359–68. PubMed Abstract
| Publisher Full Text
- 98.
Revenu C, Gilmour D:
EMT 2.0: Shaping epithelia through collective migration.
Curr Opin Genet Dev.
2009; 19(4): 338–42. PubMed Abstract
| Publisher Full Text
- 99.
Johnsson AE, Dai Y, Nobis M, et al.:
The Rac-FRET mouse reveals tight spatiotemporal control of Rac activity in primary cells and tissues.
Cell Rep.
2014; 6(6): 1153–64. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 100.
Nobis M, Herrmann D, Warren SC, et al.:
A RhoA-FRET Biosensor Mouse for Intravital Imaging in Normal Tissue Homeostasis and Disease Contexts.
Cell Rep.
2017; 21(1): 274–88. PubMed Abstract
| Publisher Full Text
- 101.
Ritsma L, Steller EJ, Beerling E, et al.:
Intravital microscopy through an abdominal imaging window reveals a pre-micrometastasis stage during liver metastasis.
Sci Transl Med.
2012; 4(158): 158ra145. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 102.
Zomer A, Maynard C, Verweij FJ, et al.:
In Vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior.
Cell.
2015; 161(5): 1046–57. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 103.
Gaggioli C, Hooper S, Hidalgo-Carcedo C, et al.:
Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells.
Nat Cell Biol.
2007; 9(12): 1392–400. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 104.
Gao MQ, Kim BG, Kang S, et al.:
Stromal fibroblasts from the interface zone of human breast carcinomas induce an epithelial-mesenchymal transition-like state in breast cancer cells in vitro.
J Cell Sci.
2010; 123(Pt 20): 3507–14. PubMed Abstract
| Publisher Full Text
- 105.
Soon PS, Kim E, Pon CK, et al.:
Breast cancer-associated fibroblasts induce epithelial-to-mesenchymal transition in breast cancer cells.
Endocr Relat Cancer.
2013; 20(1): 1–12. PubMed Abstract
| Publisher Full Text
- 106.
Yu Y, Xiao CH, Tan LD, et al.:
Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling.
Br J Cancer.
2014; 110(3): 724–32. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 107.
Ishimoto T, Miyake K, Nandi T, et al.:
Activation of Transforming Growth Factor Beta 1 Signaling in Gastric Cancer-associated Fibroblasts Increases Their Motility, via Expression of Rhomboid 5 Homolog 2, and Ability to Induce Invasiveness of Gastric Cancer Cells.
Gastroenterology.
2017; 153(1): 191–204.e16. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 108.
Itoh G, Chida S, Yanagihara K, et al.:
Cancer-associated fibroblasts induce cancer cell apoptosis that regulates invasion mode of tumours.
Oncogene.
2017; 36(31): 4434–44. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 109.
Tabassum DP, Polyak K:
Tumorigenesis: It takes a village.
Nat Rev Cancer.
2015; 15(8): 473–83. PubMed Abstract
| Publisher Full Text
- 110.
Cooks T, Pateras IS, Jenkins LM, et al.:
Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246.
Nat Commun.
2018; 9(1): 771. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 111.
Tozluoğlu M, Tournier AL, Jenkins RP, et al.:
Matrix geometry determines optimal cancer cell migration strategy and modulates response to interventions.
Nat Cell Biol.
2013; 15(7): 751–62. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 112.
Tozluoglu M, Mao Y, Bates PA, et al.:
Cost-benefit analysis of the mechanisms that enable migrating cells to sustain motility upon changes in matrix environments.
J R Soc Interface.
2015; 12(106): pii: 20141355. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 113.
Hawkins RJ, Piel M, Faure-Andre G, et al.:
Pushing off the walls: A mechanism of cell motility in confinement.
Phys Rev Lett.
2009; 102(5): 058103. PubMed Abstract
| Publisher Full Text
- 114.
Zaman MH, Kamm RD, Matsudaira P, et al.:
Computational model for cell migration in three-dimensional matrices.
Biophys J.
2005; 89(2): 1389–97. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 115.
Quail DF, Joyce JA:
Microenvironmental regulation of tumor progression and metastasis.
Nat Med.
2013; 19(11): 1423–37. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 116.
Hastings JF, Skhinas JN, Fey D, et al.:
The extracellular matrix as a key regulator of intracellular signalling networks.
Br J Pharmacol.
2018. PubMed Abstract
| Publisher Full Text
- 117.
Dumont N, Liu B, Defilippis RA, et al.:
Breast fibroblasts modulate early dissemination, tumorigenesis, and metastasis through alteration of extracellular matrix characteristics.
Neoplasia.
2013; 15(3): 249–62. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 118.
Vennin C, Herrmann D, Lucas MC, et al.:
Intravital imaging reveals new ancillary mechanisms co-opted by cancer cells to drive tumor progression [version 1; referees: 2 approved].
F1000Res.
2016; 5: pii: F1000 Faculty Rev-892. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 119.
Conway JR, Carragher NO, Timpson P:
Developments in preclinical cancer imaging: Innovating the discovery of therapeutics.
Nat Rev Cancer.
2014; 14(5): 314–28. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 120.
Vennin C, Chin VT, Warren SC, et al.:
Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis.
Sci Transl Med.
2017; 9(384): pii: eaai8504. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 121.
Laklai H, Miroshnikova YA, Pickup MW, et al.:
Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression.
Nat Med.
2016; 22(5): 497–505. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 122.
Chou A, Froio D, Nagrial AM, et al.:
Tailored first-line and second-line CDK4-targeting treatment combinations in mouse models of pancreatic cancer.
Gut.
2017; pii: gutjnl-2017-315144. PubMed Abstract
| Publisher Full Text
- 123.
Pickup MW, Laklai H, Acerbi I, et al.:
Stromally derived lysyl oxidase promotes metastasis of transforming growth factor-β-deficient mouse mammary carcinomas.
Cancer Res.
2013; 73(17): 5336–46. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 124.
Rhim AD, Oberstein PE, Thomas DH, et al.:
Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma.
Cancer Cell.
2014; 25(6): 735–47. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 125.
Özdemir BC, Pentcheva-Hoang T, Carstens JL, et al.:
Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival.
Cancer Cell.
2014; 25(6): 719–34. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 126.
Vennin C, Murphy KJ, Morton JP, et al.:
Reshaping the Tumor Stroma for Treatment of Pancreatic Cancer.
Gastroenterology.
2018; 154(4): 820–38. PubMed Abstract
| Publisher Full Text
- 127.
Cox TR, Erler JT:
Fibrosis and Cancer: Partners in Crime or Opposing Forces?
Trends Cancer.
2016; 2(6): 279–82. PubMed Abstract
| Publisher Full Text
- 128.
Mayorca-Guiliani AE, Madsen CD, Cox TR, et al.:
ISDoT: In situ decellularization of tissues for high-resolution imaging and proteomic analysis of native extracellular matrix.
Nat Med.
2017; 23(7): 890–8. PubMed Abstract
| Publisher Full Text
- 129.
Yuzhalin AE, Urbonas T, Silva MA, et al.:
A core matrisome gene signature predicts cancer outcome.
Br J Cancer.
2018; 118(3): 435–40. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 130.
Pearce OMT, Delaine-Smith RM, Maniati E, et al.:
Deconstruction of a Metastatic Tumor Microenvironment Reveals a Common Matrix Response in Human Cancers.
Cancer Discov.
2018; 8(3): 304–19. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 131.
Naba A, Clauser KR, Mani DR, et al.:
Quantitative proteomic profiling of the extracellular matrix of pancreatic islets during the angiogenic switch and insulinoma progression.
Sci Rep.
2017; 7: 40495. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 132.
Filipe EC, Chitty JL, Cox TR:
Charting the unexplored extracellular matrix in cancer.
Int J Exp Pathol.
2018; 99(2): 58–76. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 133.
Oskarsson T, Acharyya S, Zhang XH, et al.:
Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs.
Nat Med.
2011; 17(7): 867–74. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 134.
Didem T, Faruk T, Senem K, et al.:
Clinical significance of serum tenascin-c levels in epithelial ovarian cancer.
Tumour Biol.
2014; 35(7): 6777–82. PubMed Abstract
| Publisher Full Text
- 135.
Contié S, Voorzanger-Rousselot N, Litvin J, et al.:
Increased expression and serum levels of the stromal cell-secreted protein periostin in breast cancer bone metastases.
Int J Cancer.
2011; 128(2): 352–60. PubMed Abstract
| Publisher Full Text
- 136.
Keire PA, Bressler SL, Lemire JM, et al.:
A role for versican in the development of leiomyosarcoma.
J Biol Chem.
2014; 289(49): 34089–103. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 137.
Chen A, Sceneay J, Gödde N, et al.:
Intermittent hypoxia induces a metastatic phenotype in breast cancer.
Oncogene.
2018; 37(31): 4214–4225. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 138.
Deryugina EI, Kiosses WB:
Intratumoral Cancer Cell Intravasation Can Occur Independent of Invasion into the Adjacent Stroma.
Cell Rep.
2017; 19(3): 601–16. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 139.
Chang YS, di Tomaso E, McDonald DM, et al.:
Mosaic blood vessels in tumors: frequency of cancer cells in contact with flowing blood.
Proc Natl Acad Sci U S A.
2000; 97(26): 14608–13. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 140.
Khoja L, Backen A, Sloane R, et al.:
A pilot study to explore circulating tumour cells in pancreatic cancer as a novel biomarker.
Br J Cancer.
2012; 106(3): 508–16. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 141.
Schmidt H, De Angelis G, Bettendorf O, et al.:
Frequent detection and immunophenotyping of prostate-derived cell clusters in the peripheral blood of prostate cancer patients.
Int J Biol Markers.
2004; 19(2): 93–9. PubMed Abstract
| Publisher Full Text
- 142.
Krebs MG, Hou JM, Sloane R, et al.:
Analysis of circulating tumor cells in patients with non-small cell lung cancer using epithelial marker-dependent and -independent approaches.
J Thorac Oncol.
2012; 7(2): 306–15. PubMed Abstract
| Publisher Full Text
- 143.
Yu M, Bardia A, Wittner BS, et al.:
Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition.
Science.
2013; 339(6119): 580–4. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 144.
Linde N, Casanova-Acebes M, Sosa MS, et al.:
Macrophages orchestrate breast cancer early dissemination and metastasis.
Nat Commun.
2018; 9(1): 21. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 145.
Han W, Chen S, Yuan W, et al.:
Oriented collagen fibers direct tumor cell intravasation.
Proc Natl Acad Sci U S A.
2016; 113(40): 11208–13. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 146.
De Palma M, Biziato D, Petrova TV:
Microenvironmental regulation of tumour angiogenesis.
Nat Rev Cancer.
2017; 17(8): 457–74. PubMed Abstract
| Publisher Full Text
- 147.
Kerbel RS:
Tumor angiogenesis.
N Engl J Med.
2008; 358(19): 2039–49. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 148.
Kerbel R, Folkman J:
Clinical translation of angiogenesis inhibitors.
Nat Rev Cancer.
2002; 2(10): 727–39. PubMed Abstract
| Publisher Full Text
- 149.
Donnem T, Reynolds AR, Kuczynski EA, et al.:
Non-angiogenic tumours and their influence on cancer biology.
Nat Rev Cancer.
2018; 18(5): 323–36. PubMed Abstract
| Publisher Full Text
- 150.
Skobe M, Hawighorst T, Jackson DG, et al.:
Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis.
Nat Med.
2001; 7(2): 192–8. PubMed Abstract
| Publisher Full Text
- 151.
Nishida Y, Tsukushi S, Urakawa H, et al.:
High incidence of regional and in-transit lymph node metastasis in patients with alveolar rhabdomyosarcoma.
Int J Clin Oncol.
2014; 19(3): 536–43. PubMed Abstract
| Publisher Full Text
- 152.
Brown M, Assen FP, Leithner A, et al.:
Lymph node blood vessels provide exit routes for metastatic tumor cell dissemination in mice.
Science.
2018; 359(6382): 1408–11. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 153.
Huang Q, Sun Y, Ma X, et al.:
Androgen receptor increases hematogenous metastasis yet decreases lymphatic metastasis of renal cell carcinoma.
Nat Commun.
2017; 8(1): 918. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 154.
Karaman S, Detmar M:
Mechanisms of lymphatic metastasis.
J Clin Invest.
2014; 124(3): 922–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 155.
Farnsworth RH, Achen MG, Stacker SA:
The evolving role of lymphatics in cancer metastasis.
Curr Opin Immunol.
2018; 53: 64–73. PubMed Abstract
| Publisher Full Text
- 156.
Wakisaka N, Hasegawa Y, Yoshimoto S, et al.:
Primary Tumor-Secreted Lymphangiogenic Factors Induce Pre-Metastatic Lymphvascular Niche Formation at Sentinel Lymph Nodes in Oral Squamous Cell Carcinoma.
PLoS One.
2015; 10(12): e0144056. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 157.
He W, Zhong G, Jiang N, et al.:
Long noncoding RNA BLACAT2 promotes bladder cancer-associated lymphangiogenesis and lymphatic metastasis.
J Clin Invest.
2018; 128(2): 861–75. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 158.
Weichand B, Popp R, Dziumbla S, et al.:
S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1β.
J Exp Med.
2017; 214(9): 2695–713. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 159.
Yu P, Wilhelm K, Dubrac A, et al.:
FGF-dependent metabolic control of vascular development.
Nature.
2017; 545(7653): 224–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 160.
Kudo Y, Iizuka S, Yoshida M, et al.:
Periostin directly and indirectly promotes tumor lymphangiogenesis of head and neck cancer.
PLoS One.
2012; 7(8): e44488. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 161.
Le CP, Nowell CJ, Kim-Fuchs C, et al.:
Chronic stress in mice remodels lymph vasculature to promote tumour cell dissemination.
Nat Commun.
2016; 7: 10634. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 162.
Wei JC, Yang J, Liu D, et al.:
Tumor-associated Lymphatic Endothelial Cells Promote Lymphatic Metastasis By Highly Expressing and Secreting SEMA4C.
Clin Cancer Res.
2017; 23(1): 214–24. PubMed Abstract
| Publisher Full Text
- 163.
Müller A, Homey B, Soto H, et al.:
Involvement of chemokine receptors in breast cancer metastasis.
Nature.
2001; 410(6824): 50–6. PubMed Abstract
| Publisher Full Text
- 164.
Wiley HE, Gonzalez EB, Maki W, et al.:
Expression of CC chemokine receptor-7 and regional lymph node metastasis of B16 murine melanoma.
J Natl Cancer Inst.
2001; 93(21): 1638–43. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 165.
Kim M, Koh YJ, Kim KE, et al.:
CXCR4 signaling regulates metastasis of chemoresistant melanoma cells by a lymphatic metastatic niche.
Cancer Res.
2010; 70(24): 10411–21. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 166.
Meier F, Will S, Ellwanger U, et al.:
Metastatic pathways and time courses in the orderly progression of cutaneous melanoma.
Br J Dermatol.
2002; 147(1): 62–70. PubMed Abstract
| Publisher Full Text
- 167.
Aebischer D, Iolyeva M, Halin C:
The inflammatory response of lymphatic endothelium.
Angiogenesis.
2014; 17(2): 383–93. PubMed Abstract
| Publisher Full Text
- 168.
Shields JD, Kourtis IC, Tomei AA, et al.:
Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21.
Science.
2010; 328(5979): 749–52. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 169.
Lund AW, Duraes FV, Hirosue S, et al.:
VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics.
Cell Rep.
2012; 1(3): 191–9. PubMed Abstract
| Publisher Full Text
- 170.
Tewalt EF, Cohen JN, Rouhani SJ, et al.:
Lymphatic endothelial cells induce tolerance via PD-L1 and lack of costimulation leading to high-level PD-1 expression on CD8 T cells.
Blood.
2012; 120(24): 4772–82. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 171.
Olmeda D, Cerezo-Wallis D, Riveiro-Falkenbach E, et al.:
Whole-body imaging of lymphovascular niches identifies pre-metastatic roles of midkine.
Nature.
2017; 546(7660): 676–80. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 172.
Grover PK, Cummins AG, Price TJ, et al.:
Circulating tumour cells: the evolving concept and the inadequacy of their enrichment by EpCAM-based methodology for basic and clinical cancer research.
Ann Oncol.
2014; 25(8): 1506–16. PubMed Abstract
| Publisher Full Text
- 173.
Nichols AC, Lowes LE, Szeto CC, et al.:
Detection of circulating tumor cells in advanced head and neck cancer using the CellSearch system.
Head Neck.
2012; 34(10): 1440–4. PubMed Abstract
| Publisher Full Text
- 174.
O'Flaherty JD, Gray S, Richard D, et al.:
Circulating tumour cells, their role in metastasis and their clinical utility in lung cancer.
Lung Cancer.
2012; 76(1): 19–25. PubMed Abstract
| Publisher Full Text
- 175.
Takeuchi H, Kitagawa Y:
Circulating tumor cells in gastrointestinal cancer.
J Hepatobiliary Pancreat Sci.
2010; 17(5): 577–82. PubMed Abstract
| Publisher Full Text
- 176.
Iinuma H, Okinaga K, Egami H, et al.:
Usefulness and clinical significance of quantitative real-time RT-PCR to detect isolated tumor cells in the peripheral blood and tumor drainage blood of patients with colorectal cancer.
Int J Oncol.
2006; 28(2): 297–306. PubMed Abstract
| Publisher Full Text
- 177.
Cohen SJ, Punt CJ, Iannotti N, et al.:
Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer.
J Clin Oncol.
2008; 26(19): 3213–21. PubMed Abstract
| Publisher Full Text
- 178.
Cohen SJ, Punt CJ, Iannotti N, et al.:
Prognostic significance of circulating tumor cells in patients with metastatic colorectal cancer.
Ann Oncol.
2009; 20(7): 1223–9. PubMed Abstract
| Publisher Full Text
- 179.
Uen YH, Lin SR, Wu DC, et al.:
Prognostic significance of multiple molecular markers for patients with stage II colorectal cancer undergoing curative resection.
Ann Surg.
2007; 246(6): 1040–6. PubMed Abstract
| Publisher Full Text
- 180.
Swaby RF, Cristofanilli M:
Circulating tumor cells in breast cancer: a tool whose time has come of age.
BMC Med.
2011; 9: 43. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 181.
Budd GT, Cristofanilli M, Ellis MJ, et al.:
Circulating tumor cells versus imaging--predicting overall survival in metastatic breast cancer.
Clin Cancer Res.
2006; 12(21): 6403–9. PubMed Abstract
| Publisher Full Text
- 182.
Cristofanilli M, Budd GT, Ellis MJ, et al.:
Circulating tumor cells, disease progression, and survival in metastatic breast cancer.
N Engl J Med.
2004; 351(8): 781–91. PubMed Abstract
| Publisher Full Text
- 183.
Pierga JY, Hajage D, Bachelot T, et al.:
High independent prognostic and predictive value of circulating tumor cells compared with serum tumor markers in a large prospective trial in first-line chemotherapy for metastatic breast cancer patients.
Ann Oncol.
2012; 23(3): 618–24. PubMed Abstract
| Publisher Full Text
- 184.
Giuliano M, Giordano A, Jackson S, et al.:
Circulating tumor cells as prognostic and predictive markers in metastatic breast cancer patients receiving first-line systemic treatment.
Breast Cancer Res.
2011; 13(3): R67. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 185.
Miller MC, Doyle GV, Terstappen LW:
Significance of Circulating Tumor Cells Detected by the CellSearch System in Patients with Metastatic Breast Colorectal and Prostate Cancer.
J Oncol.
2010; 2010: 617421. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 186.
Cristofanilli M, Hayes DF, Budd GT, et al.:
Circulating tumor cells: a novel prognostic factor for newly diagnosed metastatic breast cancer.
J Clin Oncol.
2005; 23(7): 1420–30. PubMed Abstract
| Publisher Full Text
- 187.
Ignatiadis M, Kallergi G, Ntoulia M, et al.:
Prognostic value of the molecular detection of circulating tumor cells using a multimarker reverse transcription-PCR assay for cytokeratin 19, mammaglobin A, and HER2 in early breast cancer.
Clin Cancer Res.
2008; 14(9): 2593–600. PubMed Abstract
| Publisher Full Text
- 188.
Dasgupta A, Lim AR, Ghajar CM:
Circulating and disseminated tumor cells: harbingers or initiators of metastasis?
Mol Oncol.
2017; 11(1): 40–61. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 189.
Alix-Panabières C, Pantel K:
Challenges in circulating tumour cell research.
Nat Rev Cancer.
2014; 14(9): 623–31. PubMed Abstract
| Publisher Full Text
- 190.
Pantel K, Speicher MR:
The biology of circulating tumor cells.
Oncogene.
2016; 35(10): 1216–24. PubMed Abstract
| Publisher Full Text
- 191.
Aceto N, Toner M, Maheswaran S, et al.:
En Route to Metastasis: Circulating Tumor Cell Clusters and Epithelial-to-Mesenchymal Transition.
Trends Cancer.
2015; 1(1): 44–52. PubMed Abstract
| Publisher Full Text
- 192.
Douma S, van Laar T, Zevenhoven J, et al.:
Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB.
Nature.
2004; 430(7003): 1034–9. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 193.
Yu M, Ting DT, Stott SL, et al.:
RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis.
Nature.
2012; 487(7408): 510–3. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 194.
Labelle M, Hynes RO:
The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination.
Cancer Discov.
2012; 2(12): 1091–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 195.
Labelle M, Begum S, Hynes RO:
Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis.
Cancer Cell.
2011; 20(5): 576–90. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 196.
Palumbo JS, Talmage KE, Massari JV, et al.:
Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells.
Blood.
2005; 105(1): 178–85. PubMed Abstract
| Publisher Full Text
- 197.
Palumbo JS, Talmage KE, Massari JV, et al.:
Tumor cell-associated tissue factor and circulating hemostatic factors cooperate to increase metastatic potential through natural killer cell-dependent and-independent mechanisms.
Blood.
2007; 110(1): 133–41. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 198.
Duda DG, Duyverman AM, Kohno M, et al.:
Malignant cells facilitate lung metastasis by bringing their own soil.
Proc Natl Acad Sci U S A.
2010; 107(50): 21677–82. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 199.
Faltas B:
Targeting hematogenous spread of circulating tumor cells by a chemotactic drug-eluting IVC filter to prevent pulmonary and systemic metastasis.
Med Hypotheses.
2010; 74(4): 668–9. PubMed Abstract
| Publisher Full Text
- 200.
Wirtz D, Konstantopoulos K, Searson PC:
The physics of cancer: the role of physical interactions and mechanical forces in metastasis.
Nat Rev Cancer.
2011; 11(7): 512–22. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 201.
Barnes JM, Nauseef JT, Henry MD:
Resistance to fluid shear stress is a conserved biophysical property of malignant cells.
PLoS One.
2012; 7(12): e50973. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 202.
Mitchell MJ, King MR:
Computational and experimental models of cancer cell response to fluid shear stress.
Front Oncol.
2013; 3: 44. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 203.
Northcott JM, Dean IS, Mouw JK, et al.:
Feeling Stress: The Mechanics of Cancer Progression and Aggression.
Front Cell Dev Biol.
2018; 6: 17. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 204.
Headley MB, Bins A, Nip A, et al.:
Visualization of immediate immune responses to pioneer metastatic cells in the lung.
Nature.
2016; 531(7595): 513–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 205.
Fidler IJ, Nicolson GL:
Organ selectivity for implantation survival and growth of B16 melanoma variant tumor lines.
J Natl Cancer Inst.
1976; 57(5): 1199–202. PubMed Abstract
| Publisher Full Text
- 206.
Hart IR, Fidler IJ:
Role of organ selectivity in the determination of metastatic patterns of B16 melanoma.
Cancer Res.
1980; 40(7): 2281–7. PubMed Abstract
- 207.
Au SH, Storey BD, Moore JC, et al.:
Clusters of circulating tumor cells traverse capillary-sized vessels.
Proc Natl Acad Sci U S A.
2016; 113(18): 4947–52. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 208.
Schumacher D, Strilic B, Sivaraj KK, et al.:
Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor.
Cancer Cell.
2013; 24(1): 130–7. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 209.
Honda M, Kubes P:
Neutrophils and neutrophil extracellular traps in the liver and gastrointestinal system.
Nat Rev Gastroenterol Hepatol.
2018; 15(4): 206–21. PubMed Abstract
| Publisher Full Text
- 210.
Park J, Wysocki RW, Amoozgar Z, et al.:
Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps.
Sci Transl Med.
2016; 8(361): 361ra138. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 211.
Wolf MJ, Hoos A, Bauer J, et al.:
Endothelial CCR2 signaling induced by colon carcinoma cells enables extravasation via the JAK2-Stat5 and p38MAPK pathway.
Cancer Cell.
2012; 22(1): 91–105. PubMed Abstract
| Publisher Full Text
- 212.
Qian BZ, Li J, Zhang H, et al.:
CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis.
Nature.
2011; 475(7355): 222–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 213.
Harney AS, Arwert EN, Entenberg D, et al.:
Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA.
Cancer Discov.
2015; 5(9): 932–43. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 214.
Hiratsuka S, Ishibashi S, Tomita T, et al.:
Primary tumours modulate innate immune signalling to create pre-metastatic vascular hyperpermeability foci.
Nat Commun.
2013; 4: 1853. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 215.
Zhou W, Fong MY, Min Y, et al.:
Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis.
Cancer Cell.
2014; 25(4): 501–15. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 216.
Gupta GP, Nguyen DX, Chiang AC, et al.:
Mediators of vascular remodelling co-opted for sequential steps in lung metastasis.
Nature.
2007; 446(7137): 765–70. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 217.
Padua D, Zhang XH, Wang Q, et al.:
TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4.
Cell.
2008; 133(1): 66–77. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 218.
Huang Y, Song N, Ding Y, et al.:
Pulmonary vascular destabilization in the premetastatic phase facilitates lung metastasis.
Cancer Res.
2009; 69(19): 7529–37. PubMed Abstract
| Publisher Full Text
- 219.
Ahirwar DK, Nasser MW, Ouseph MM, et al.:
Fibroblast-derived CXCL12 promotes breast cancer metastasis by facilitating tumor cell intravasation.
Oncogene.
2018; 37: 4428–4442. PubMed Abstract
| Publisher Full Text
- 220.
Peinado H, Zhang H, Matei IR, et al.:
Pre-metastatic niches: organ-specific homes for metastases.
Nat Rev Cancer.
2017; 17(5): 302–17. PubMed Abstract
| Publisher Full Text
- 221.
Strilic B, Yang L, Albarrán-Juárez J, et al.:
Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis.
Nature.
2016; 536(7615): 215–8. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 222.
Huh SJ, Liang S, Sharma A, et al.:
Transiently entrapped circulating tumor cells interact with neutrophils to facilitate lung metastasis development.
Cancer Res.
2010; 70(14): 6071–82. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 223.
Spicer JD, McDonald B, Cools-Lartigue JJ, et al.:
Neutrophils promote liver metastasis via Mac-1-mediated interactions with circulating tumor cells.
Cancer Res.
2012; 72(16): 3919–27. PubMed Abstract
| Publisher Full Text
- 224.
Spiegel A, Brooks MW, Houshyar S, et al.:
Neutrophils Suppress Intraluminal NK Cell-Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells.
Cancer Discov.
2016; 6(6): 630–49. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 225.
Coffelt SB, Kersten K, Doornebal CW, et al.:
IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis.
Nature.
2015; 522(7556): 345–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 226.
Follain G, Osmani N, Azevedo AS, et al.:
Hemodynamic Forces Tune the Arrest, Adhesion, and Extravasation of Circulating Tumor Cells.
Dev Cell.
2018; 45(1): 33–52.e12. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 227.
Ghajar CM:
Metastasis prevention by targeting the dormant niche.
Nat Rev Cancer.
2015; 15(4): 238–47. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 228.
Paget S:
The distribution of secondary growths in cancer of the breast. 1889.
Cancer Metastasis Rev.
1989; 8(2): 98–101. PubMed Abstract
- 229.
Malanchi I, Santamaria-Martínez A, Susanto E, et al.:
Interactions between cancer stem cells and their niche govern metastatic colonization.
Nature.
2011; 481(7379): 85–9. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 230.
Clever D, Roychoudhuri R, Constantinides MG, et al.:
Oxygen Sensing by T Cells Establishes an Immunologically Tolerant Metastatic Niche.
Cell.
2016; 166(5): 1117–1131.e14. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 231.
Shibue T, Weinberg RA:
Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs.
Proc Natl Acad Sci U S A.
2009; 106(25): 10290–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 232.
Shibue T, Brooks MW, Inan MF, et al.:
The outgrowth of micrometastases is enabled by the formation of filopodium-like protrusions.
Cancer Discov.
2012; 2(8): 706–21. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 233.
El Touny LH, Vieira A, Mendoza A, et al.:
Combined SFK/MEK inhibition prevents metastatic outgrowth of dormant tumor cells.
J Clin Invest.
2014; 124(1): 156–68. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 234.
Croucher DR, Saunders DN, Lobov S, et al.:
Revisiting the biological roles of PAI2 (SERPINB2) in cancer.
Nat Rev Cancer.
2008; 8(7): 535–45. PubMed Abstract
| Publisher Full Text
- 235.
Valiente M, Obenauf AC, Jin X, et al.:
Serpins promote cancer cell survival and vascular co-option in brain metastasis.
Cell.
2014; 156(5): 1002–16. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 236.
Harris NLE, Vennin C, Conway JRW, et al.:
SerpinB2 regulates stromal remodelling and local invasion in pancreatic cancer.
Oncogene.
2017; 36(30): 4288–98. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 237.
Neman J, Termini J, Wilczynski S, et al.:
Human breast cancer metastases to the brain display GABAergic properties in the neural niche.
Proc Natl Acad Sci U S A.
2014; 111(3): 984–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 238.
Weilbaecher KN, Guise TA, McCauley LK:
Cancer to bone: a fatal attraction.
Nat Rev Cancer.
2011; 11(6): 411–25. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 239.
Sleeman JP:
The metastatic niche and stromal progression.
Cancer Metastasis Rev.
2012; 31(3–4): 429–40. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 240.
Kaplan RN, Riba RD, Zacharoulis S, et al.:
VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche.
Nature.
2005; 438(7069): 820–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 241.
Hiratsuka S, Nakamura K, Iwai S, et al.:
MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis.
Cancer Cell.
2002; 2(4): 289–300. PubMed Abstract
| Publisher Full Text
- 242.
Cox TR, Rumney RMH, Schoof EM, et al.:
The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase.
Nature.
2015; 522(7554): 106–10. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 243.
Costa-Silva B, Aiello NM, Ocean AJ, et al.:
Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver.
Nat Cell Biol.
2015; 17(6): 816–26. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 244.
Hoshino A, Costa-Silva B, Shen TL, et al.:
Tumour exosome integrins determine organotropic metastasis.
Nature.
2015; 527(7578): 329–35. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 245.
Peinado H, Alečković M, Lavotshkin S, et al.:
Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET.
Nat Med.
2012; 18(6): 883–91. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 246.
Sánchez CA, Andahur EI, Valenzuela R, et al.:
Exosomes from bulk and stem cells from human prostate cancer have a differential microRNA content that contributes cooperatively over local and pre-metastatic niche.
Oncotarget.
2016; 7(4): 3993–4008. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 247.
Aguado BA, Bushnell GG, Rao SS, et al.:
Engineering the pre-metastatic niche.
Nat Biomed Eng.
2017; 1: pii: 0077. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 248.
Aguado BA, Caffe JR, Nanavati D, et al.:
Extracellular matrix mediators of metastatic cell colonization characterized using scaffold mimics of the pre-metastatic niche.
Acta Biomater.
2016; 33: 13–24. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 249.
Janni W, Vogl FD, Wiedswang G, et al.:
Persistence of disseminated tumor cells in the bone marrow of breast cancer patients predicts increased risk for relapse--a European pooled analysis.
Clin Cancer Res.
2011; 17(9): 2967–76. PubMed Abstract
| Publisher Full Text
- 250.
Suzuki M, Mose ES, Montel V, et al.:
Dormant cancer cells retrieved from metastasis-free organs regain tumorigenic and metastatic potency.
Am J Pathol.
2006; 169(2): 673–81. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 251.
Malladi S, Macalinao DG, Jin X, et al.:
Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT.
Cell.
2016; 165(1): 45–60. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 252.
Koebel CM, Vermi W, Swann JB, et al.:
Adaptive immunity maintains occult cancer in an equilibrium state.
Nature.
2007; 450(7171): 903–7. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 253.
Müller-Hermelink N, Braumüller H, Pichler B, et al.:
TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis.
Cancer Cell.
2008; 13(6): 507–18. PubMed Abstract
| Publisher Full Text
- 254.
Eyles J, Puaux AL, Wang X, et al.:
Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma.
J Clin Invest.
2010; 120(6): 2030–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 255.
Bui AT, Laurent F, Havard M, et al.:
SMAD signaling and redox imbalance cooperate to induce prostate cancer cell dormancy.
Cell Cycle.
2015; 14(8): 1218–31. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 256.
Gao H, Chakraborty G, Lee-Lim AP, et al.:
The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites.
Cell.
2012; 150(4): 764–79. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 257.
Zhang XH, Wang Q, Gerald W, et al.:
Latent bone metastasis in breast cancer tied to Src-dependent survival signals.
Cancer Cell.
2009; 16(1): 67–78. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 258.
Bragado P, Estrada Y, Parikh F, et al.:
TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling.
Nat Cell Biol.
2013; 15(11): 1351–61. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 259.
Sharma S, Xing F, Liu Y, et al.:
Secreted Protein Acidic and Rich in Cysteine (SPARC) Mediates Metastatic Dormancy of Prostate Cancer in Bone.
J Biol Chem.
2016; 291(37): 19351–63. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 260.
Kobayashi A, Okuda H, Xing F, et al.:
Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone.
J Exp Med.
2011; 208(13): 2641–55. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 261.
Ghajar CM, Peinado H, Mori H, et al.:
The perivascular niche regulates breast tumour dormancy.
Nat Cell Biol.
2013; 15(7): 807–17. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 262.
De Cock JM, Shibue T, Dongre A, et al.:
Inflammation Triggers Zeb1-Dependent Escape from Tumor Latency.
Cancer Res.
2016; 76(23): 6778–84. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 263.
Barkan D, El Touny LH, Michalowski AM, et al.:
Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment.
Cancer Res.
2010; 70(14): 5706–16. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 264.
Lawson MA, McDonald MM, Kovacic N, et al.:
Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche.
Nat Commun.
2015; 6: 8983. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 265.
Gandalovičová A, Rosel D, Fernandes M, et al.:
Migrastatics-Anti-metastatic and Anti-invasion Drugs: Promises and Challenges.
Trends Cancer.
2017; 3(6): 391–406. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 266.
Sporn MB:
The war on cancer.
Lancet.
1996; 347(9012): 1377–81. PubMed Abstract
| Publisher Full Text
- 267.
Karagiannis GS, Pastoriza JM, Wang Y, et al.:
Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism.
Sci Transl Med.
2017; 9(397): pii: eaan0026. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
Comments on this article Comments (0)